
How to Prepare for Your Post-Pandemic On-Site GMP Regulatory Inspection?
How can you best prepare for a post-pandemic on-site GMP regulatory inspection, as in-person inspections return around the world?
John Johnson, Vice President in our Health Sciences division, recently presented on the issue of preparing for your post-pandemic on-site GMP regulatory inspection to the Research & Quality Association (RQA) Ireland forum. The webinar looked at:
- How the events of 2020 and 2021 have altered the priorities when preparing for, and executing, a GMP regulatory inspection
- The need to modify the approach to site preparedness
- The hot topics in the sector from now until the end of 2022
- A model approach to tackling the difficult questions ahead
- Best practices in preparing an effective and timely CAPA or GMP remediation plan
Watch the Webinar

We have all witnessed the effects of COVID-19 and government policy in the past two years. However, have we stopped to consider how they have impacted the pharma industry? Our conversations with clients and industry peers have shown that some firms are thriving. Conversely, issues like supply chain disruptions, with delays in the delivery of excipients, APIs and other raw materials, have had a negative impact on many firms. These supply chain issues have, inevitably, led to drug shortages across the industry. We are also seeing a deficit in the availability of talent, with firms struggling to fill vacancies. This issue has placed a real stress on the industry. Supply chain shortages and shortages of available talent lead companies to make risk-based decisions that may not normally be taken. Also, CAPEX investments that many companies have planned have been put on hold.
How Have These Events Affected Some Firms’ Ability to Meet cGMP?
Firstly, legacy facilities are now two years older and may not have been upgraded. In that instance, there is a need to ask if the facility still complies with cGMP. The new Eudralex Vol. 4 Annex 1 will be introduced soon, and companies need to consider if their legacy sterile facility is still compliant with what is included in Annex 1.
Conversations with companies across the world have shown that scheduled adherence to the Pharmaceutical Quality System (PQS) has really suffered because of COVID-19 restrictions on businesses. We have seen that the engagement in deviation investigation reports has suffered and that the completion of those reports within 30 days has been adversely impacted. This then has a knock-on effect on the target completion dates of the CAPA.
Other reports in the quality system that are now either parked or late include:
- Qualification and validation reports
- Annual product review
- Change control
- Vendor appraisal
- Internal and external audits
- Planned preventive maintenance, corrective maintenance and calibration
Many, if not all, of these reports have been parked with some justification. Being able to justify to regulatory inspectors why the reports have been parked is vitally important, as regulators will be keen to understand why backlogs in documentation exist.
Supply chain issues, staff shortages, documentation backlogs and more can drive companies to make value judgments and compromises that may not normally even be contemplated. Ultimately, this can lead to subjective decisions that result in pain points for the business and risks to product quality.
Typical Life Cycle When Experiencing Issues
- Discovery
- Disbelief, denial and anger
- Stasis
- Acceptance
- Evaluation
- Planning
- Execution of actions
- Effectiveness checks
What Is Happening From a Regulatory Agency Point of View?
The most important impact of the events of the past two years has been the reduction in the number of inspections happening. This backlog has recently been highlighted by events in the United States, where the Committee on Energy and Commerce of the U.S. Congress wrote to the FDA, seeking information on the measures the FDA was taking to reduce the backlog of pharmaceutical manufacturing facility inspections. This shows there is a desire in the industry and from regulators to get back to face-to-face inspections as soon as is practical. Our colleagues, in conversation with regulators and client companies, are expecting to see a ramping up of activity in this regard in the final quarter of 2021. The anticipated result is that there will be increased regulatory enforcement as companies begin to realise that some of the decisions that have had to be made were not easily justified.
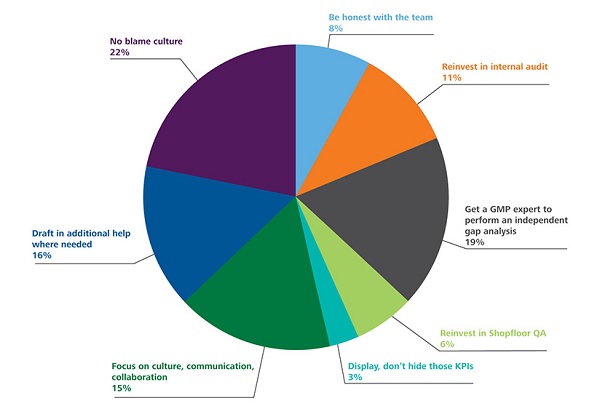
Poll Results: How Would You Modify Your Approach to GMP Inspection Preparedness Post Pandemic?
We conducted a poll of experts working in the pharmaceutical industry and asked them to identify the immediate steps in addressing post-COVID-19 GMP deficiencies across the pharma quality system. The results are below:
Six Critical Steps to Getting Back in Control
1. Own the fact that there is an issue
2. Be realistic and use existing resource as best as possible
3. Action taken must be based on evidence
4. Focus on the key risks, i.e., the “critical few”:
a) Adulteration
b) Misbranding
c) Data integrity
5. Use a project charter
6. Keep going and make time to check in regularly
When trying to verify if the issues are fixed, we always recommend using a process called “In Place, in Use and in Control”:
What Are the “Hot Topics”?
These are what we see as the “hot topics” that are likely to come up in your next inspection, utilising the six systems defined in the FDA’s quality system inspection technique approach (Drug Manufacturing Inspections CPGM 7356.002).
Materials Control
Short-dated or expired materials or components will be one of the post-pandemic hot topics. Compliance with incoming QC checks and inspection, along with supplier approval and vendor assurance, will also feature in this part of any discussion on materials control.
Manufacturing Control
The management and possible overstretching of key assets will be looked at in this section. Here we are not just suggesting that facilities, utilities and equipment assets will be considered. We believe that human assets will also be considered. Regulators will be looking at what role human error will play here at a time when companies have had capacity and other constraints.
Packaging and Labeling Control
Regulators will be keen to understand what issues have occurred with nonstandard labelling and printing concerns. They will also seek proof that people have adhered to in-process checks and examine whether there have been issues with product changeovers.
Facilities and Equipment Control
The condition and the ability of facilities to comply with cGMP will be the central topic in this instance. At a time when there has been reduced availability of spare parts and machinery, will the change management system be able to confirm that like-for-like changes have always been a priority?
Quality Control
Regulators will be very interested in the prevalence and engagement of terminated analyses, which signify an error. Also, the pressures on a business to deal with staffing shortages rear their head again in quality control. Truncated training and onboarding may see an increase in the prevalence of out-of-specification results. Finally, for now, the ability to demonstrate data integrity from start to finish in the manufacturing process is integral and will be investigated by regulators.
Quality Management
In this instance, issues around backlogs, shop floor quality assurance and oversight of processes from the quality team will be central to the questioning of regulators taking part in a post-pandemic investigation.
How to Deal With Difficult Questions
- Be prepared
- Do not hide in the shadows
- Assess risks using the RPN = S x O x D from ICH Q9
- Find all potential root causes
- Treat each issue as an opportunity to learn and a vector for driving down severity, occurrence or ambiguity.
Consider Developing a “Most Feared Question” Playbook
Many firms adopt the approach of building a playbook of questions they would least like to receive and collating it into a document to be used for training purposes. Most feared questions require answers, and the NSF approach to building a playbook is to:
- Acknowledge the issue and define it well
- Refer to cGMPs
- Summarize the current SOP or the context of the issue
- Emphasize the present performance of the current system
- Consider the chain of risk when looking at product quality impact assessment. Each risk must be well defined, unambiguous and have a specified start and end date
- Summarize any planned improvements, effectiveness checks and measures
- Summarize the project charter
One final tip: Do not try to defend the indefensible.
Handling the End of the Inspection
At the end of the inspection, it is likely that issues will have been identified and that you will have to create a CAPA or regulatory response. We have developed a structured approach to dealing with these issues.
We Can Help
The regulatory landscape has changed hugely in 2020 and 2021. It will change again as the number of in-person inspections starts increasing. We always advise companies to keep an eye on the big-impact items and not to allow themselves to get swamped.
Our team of ex-regulators and experienced industry experts can help your company to prepare for an inspection or deal with issues that arise from an inspection. If you would like us to assist, just complete the form below.









