
Artificial Intelligence - A current and future tool to learn chemical sciences
Ruchi Bharti, Department of Chemistry, University Institute of Sciences, Chandigarh University
Chemists are increasingly employing artificial intelligence (AI) to accomplish diverse lists of tasks. Initially, the need to enhance the drug development process and lower its massive development costs and time pushed the AI or computer-based research in chemistry. In this article, we have described how computer-based intelligence is utilised in chemistry to enhance knowledge, which further helps to do more creative or innovative research in minimal time.
1. Introduction
Artificial Intelligence (AI) is used in various tasks, such as human speech recognition, creating strategic games, autonomously operating cars, chemistry, etc. Artificial intelligence and machine learning have demonstrated their potential role in predicting chemistry and synthetic planning of small molecules; at least a few reports of companies employing in silico synthetic planning in their overall approach to accessing target molecules. In chemistry, data has a significant role; the accumulated data is interpreted, and the knowledge/findings are derived. Researchers and scientists conduct many experiments focused on the chemistry of molecules or atoms and collect data. The collected data is then used to study atoms or molecules' chemical and physical properties, reactivity in chemical reactions, or biological activity. Also, the collected data were used by analogy to make predictions or derive models for the principles underlying the data. This article attempts to show how chemoinformatics methods have been instrumental in using chemistry-related data to enhance our knowledge and how that knowledge can be used to accelerate chemical innovation.[1]
2. Role of AI in Learning in Chemistry
Due to the experiments in chemistry on a considerable scale, a massive increase in chemical data can be seen. Fortunately, along with this growth, computer-based technology has also advanced and is becoming more powerful day by day. Due to the advancement of computers, it is being used to solve mathematical operations in the equations of quantum mechanics. It gives us more convenience in accurately doing the complex quantum mechanics calculations of chemical and physical data. However, this type of learning is considered Deductive Learning. In this type of learning, data is produced using predefined information. However, computers are used to perform logical operations, so if we develop software that can convert data into information and use that information to enhance knowledge, then the properties of any molecules can be predicted. This type of learning is known as Inductive Learning. In this, data obtained is used to give information. Figure 1 represents Deductive and inductive learning. [1]
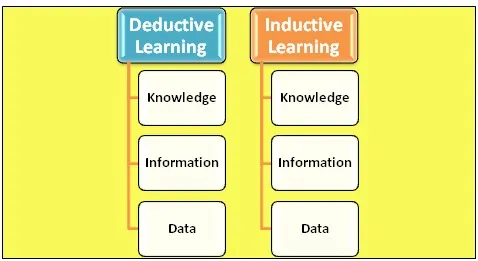
Figure 1: Deductive and Inductive learning.[1]
2.1. Cheminformatics
In the 1960s, some computer methods were developed based on inductive learning, which later became the field of chemoinformatics.[2, 3] Chemininformatics, also known as chemical informatics, was given in 1998 by F.K. Brown.[4] The field of cheminformatics, using information technology and computer science, has helped solve a wide range of chemistry-related problems. It deals with statistics, discrete mathematics, information and computation theory, soft computing, artificial intelligence, web technology, database and information systems, algorithms, etc., to generate new information, which is further used to enhance the knowledge of chemistry.
2.1.1. Cheminformatics in Databases generation and prediction of 3D structures of the molecules.
In today's time, the development of computer technology is seen as speedy growth. Various chemical structures and reactions are processed, and the databases are prepared and stored. Also, the chemical structures can be efficiently coded so that their various desirable possibilities can be processed and manipulated. Usually, to represent the structure of various molecules, it has become a rule to represent by a connection table. The list of atoms and bonds is present in the connection table, which provides information about the molecule. The most commonly used code is SMILES.[5] SMILES can be easily converted into a connection table, and information about molecules can be easily shared on the World Wide Internet. Many databases are seen today, which communicate the information of molecules with computers in the language of chemists.
Some of them are listed below:
• Cambridge Structural Database (CSD): Cambridge Structural Database (CSD) contains information on over 1 million molecules.[6] This database contains only those organic and organometallic compounds whose 3D structure has been experimentally determined. However, the number is significantly less. In 1990, there were 22,000,000 molecules in the Chemical Abstracts Service Registry System and 230,000 structures in the CSD. With the help of 230,000 known structures present in CSD, a procedure was devised named CORINA. With the help of this procedure, an attempt was made to predict the 3-dimensional structures of molecules. Surprisingly, the accuracy of these results was more than 99 per cent.[7]
• Chemical Abstracts Service Registry System:
Structures of more than 160 million inorganic and organic molecules are present in the database of the CAS Registry System. More than 68 million biosequences are present in its database [8], which can help study the interaction between molecules and various proteins via computer-assisted molecular docking and molecular simulations).
• Reaxys:
The Reaxys database is derived from the Patent Chemistry, Gmelin and Beilstein databases. More than 500 million experimental properties (chemical and physical) in this database.[9]
Researchers have collected different types of chemistry-related data in large amounts, which are present in these databases and require different computational methods. Modern chemical science cannot even be imagined without these databases. Cheminformatics plays a very crucial role in modern chemistry.
2.1.2. Cheminformatics and Drug Discovery
Outbreaks of various types of diseases have been seen for the last few years, so more emphasis is given to synthesizing such molecules with bioactivity. For such purposes, combinatorial synthesis can be beneficial in producing a series of new molecules in a short time. However, it also has some limitations. For example, all the molecules of series do not have bioactivity. Hence, to avoid wastage of chemicals and time, we can predict which molecules will be bioactive, and only those molecules should be synthesized using cheminformatics. With the increasing competition and cost of drug design, bioactive molecules are being manufactured using tailor-made designs rather than hit or trial methodologies. Using tailor-made designs helps make target-based drugs and reduces the time taken for drug development. In an earlier time, more than a year time was required to screen one lakh individual molecules against drug targets [10, 11]. However, in present times, by utilising the applications of cheminformatics, thousands of molecules can be screened per day, and further clinical trials can be performed. Figure 2 represents how cheminformatics plays an essential role in drug discovery.
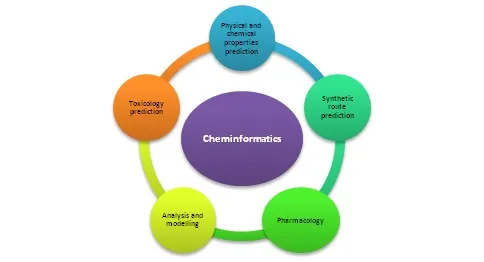
Figure 2: Utilisation of cheminformatics in various predictions and studies.
2.1.3 Reaction Prediction
The most significant task of researchers is to predict in advance what the product of any reaction can be. The synthetic root of any reaction can be predicted by using computational quantum mechanics. Transition states can be calculated with the help of the methods of quantum mechanics. However, it is difficult in today's time to calculate the effect of solvent and temperature on the reaction route. Databases can prove to be a perfect option to solve this problem. In the database, the reaction which will be of interest or the reaction related to it can be searched and valuable information can be obtained, which can be further utilised to synthesize the desired molecule.
2.1.4. Computer-Assisted Structure Elucidation (CASE)
Recently, methods were developed automatically collect the data and predict the structure using spectroscopic techniques. Two groups from Japan and USA worked together on such methods in this regard.[12, 13] Based on this report, the SESAME and CHEMICS system was used for a long time, and its results were also good, but they were not used so much at the global level. Elyashberg also worked on all the advances in NMR and created a system that got excellent results in describing the structure. In today's time, it is commercially available in the name of ACD / LAB.[14-16]
2.1.5. Cheminformatics and Bioinformatics in Biochemical pathways prediction
Databases prove to be most helpful in finding out the chemical reactions that occur in the living species and what are the pathways that reaction follows. BioPath.explore [17, 18] is among those databases that store the reaction and pathway-related information. Information from databases is being combined with the chemical knowledge to create systems that can mimic the biochemical pathways so that the natural organic compounds can be easily synthesized in the lab.
2.1.6. Cheminformatics in Predicting Absorption, Distribution, Metabolism, and Excretion (ADME) properties
Whenever a drug is manufactured, it has a particular path in the body. To understand this, we take an example of an oral drug. If each molecule manually starts studying experimentally, then it will take many months, due to which the cost of studying their properties will also increase with time. In most of cases, the main reason for the failure of any drug in clinical trials is its poor ADME properties.[19] At the time of drug discovery, when designing a molecule, if at the same time the ADME properties of that molecule are studied, then the time and chemical wastage can be reduced. The field of cheminformatics is found to be very useful in solving ADME-related problems. With time, the cheminformatics field has evolved a lot. modem.ucsd.edu [20] is a cheminformatic-based website used to predict ADME properties worldwide. modem.ucsd.edu uses the following databases:
• Human oral bioavailability database.
• Human oral absorption database.
• logB database.
• Caco-2 permeability database.
• logS database.
• logD database.
• pKa database.
• logP database.
Using abovementioned databases they effectively predict the following properties:
• Human oral availability.
• CYP450 metabolism.
• The volume of distribution.
• Human plasma protein binding.
• P-GP inhibition
• Blood-brain barrier permeability.
• Human intestinal absorption.
• Aqueous solubility.
• Octanol-water distribution coefficient.
• Octanol-water partition coefficient.
Predicted results obtained can be beneficial in studying the different aspects of the drug molecules and is very helpful in the research related to drug discovery.
Conclusion:
Machine learning-based techniques can now assist scientists in designing and synthesising novel compounds with bioactivities. In many circumstances, these tools perform excellently and increase chemist output. In the coming years, we should expect researchers to make significant improvements, and these tools will become more essential to chemists.
Reference
[1] Gasteiger, J., Chemistry in times of artificial intelligence. ChemPhysChem 2020, 21 (20), 2233-2242.
[2] Engel, T.; Gasteiger, J., Chemoinformatics: basic concepts and methods. John Wiley & Sons: 2018.
[3] Gasteiger, J.; Engel, T., Applied Chemoinformatics: Achievements and Future Opportunities. Wiley-VCH Verlag GmBH: 2018.
[4] Brown, F. K., Chapter 35 - Chemoinformatics: What is it and How does it Impact Drug Discovery. In Annual Reports in Medicinal Chemistry, Bristol, J. A., Ed. Academic Press: 1998; Vol. 33, pp 375-384.
[5] Weininger, D., SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. Journal of chemical information and computer sciences 1988, 28 (1), 31-36.
[6] https://www.ccdc.cam.ac.uk/solutions/csd-system/components/csd/, last accessed May 17, 2022.
[7] Sadowski, J.; Gasteiger, J., From atoms and bonds to three-dimensional atomic coordinates: automatic model builders. Chemical Reviews 1993, 93 (7), 2567-2581.
[8] https://www.cas.org/support/documentation/chemical-substances, last accessed May 17, 2022.
[9] https://www.elsevier.com/solutions/reaxys, last accessed May 17, 2022.
[10] Gallop, M. A.; Barrett, R. W.; Dower, W. J.; Fodor, S. P. A.; Gordon, E. M., Applications of combinatorial technologies to drug discovery. 1. Background and peptide combinatorial libraries. Journal of medicinal chemistry 1994, 37 (9), 1233-1251.
[11] Hecht, P., High-throughput screening: beating the odds with informatics-driven chemistry. Curr. Drug Discov 2002, 21-24.
[12] Munk, M. E.; Sodano, C. S.; McLean, R. L.; Haskell, T. H., Actinobolin. I. Structure of actinobolamine. Journal of the American Chemical Society 1967, 89 (16), 4158-4165.
[13] Sasaki, S.; Abe, H.; Ouki, T.; Sakamoto, M.; Ochiai, S., Automated structure elucidation of several kinds of aliphatic and alicyclic compounds. Analytical Chemistry 1968, 40 (14), 2220-2223.
[14] Elyashberg, M.; Blinov, K.; Molodtsov, S.; Smurnyy, Y.; Williams, A. J.; Churanova, T., Computer-assisted methods for molecular structure elucidation: realising a spectroscopist's dream. Journal of Cheminformatics 2009, 1 (1), 1-26.
[15] Elyashberg, M.; Williams, A. J., Computer-based structure elucidation from spectral data. Springer: 2015; Vol. 89.
[16] https://www.acdlabs.com/products/com_iden/elucidation/struc_eluc/, last accessed May 17, 2022.
[17] Reitz, M.; Sacher, O.; Tarkhov, A.; Trümbach, D.; Gasteiger, J., Enabling the exploration of biochemical pathways. Organic & biomolecular chemistry 2004, 2 (22), 3226-3237.
[18] https://v12.chemtunes.com/biopath3/, last accessed May 16, 2022.
[19] ADME - TDC (tdcommons.ai) last accessed May 16, 2022.
[20] ADME predictions (ucsd.edu) last accessed May 16, 2022.

Ruchi Bharti received her M.Sc. from Patna University, in organic chemistry. PhD from National Institute of Technology Patna (NIT Patna), in 2012. There she worked on the development of new organocatalysts and their utilization in the synthesis of diverse heterocyclic compounds via multimulticomponent reactions. In 2017, she enrolled as assistant professor at R. P Sharma Institute of Technology, Since 2018, she joined Department of Chemistry, University Institute of Sciences, Chandigarh University, Punjab as Assistant Professor. Her recent research work is focused on the functionalisation of diverse heterocycles, multicomponent reactions, organocatalysis and their related biological and medicinal applications.




