Changing Paradigms in Use of AI in Chemical Sciences
Prashant S. Kharkar, Department of Pharmaceutical Sciences and Technology, Institute of Chemical Technology
Akash S. Sirsat, Department of Pharmaceutical Sciences and Technology, Institute of Chemical Technology
Chemical Sciences have undergone a significant metamorphosis in recent years, driven by modern chemical methods, state-of-the-art instrumentation, and high-end computational power. Notably, the use of sophisticated AI/ML tools is increasing across various applications within the field. The present article outlines major developments in AI/ML and discusses their impact on current chemistry practices, with particular emphasis on drug discovery and allied fields.
Introduction
During my postgraduate studies three decades ago, I was drawn to the emerging fields of cheminformatics and computer-aided molecular design (CAMD). Working with Silicon Graphics workstations and advanced computational tools of that era significantly shaped my scientific direction. Throughout my career, I have explored 2D and 3D QSAR, de novo molecular design, and early predictive models for pharmacokinetics and toxicity [1]. At that time, these areas were still developing due to limited and unreliable datasets. It has been remarkable to witness the evolution of the tools we now use daily, many of which were then just being built and debated.
Computational limitations were a constant reality. Simulations ran in batches, results were pieced together manually, and processing power was scarce by today’s standards. I vividly remember reading that simulating the folding of a 100-amino-acid protein could take a thousand years on the fastest computers available, a figure that seemed almost absurd then. Looking back, it feels less like an exaggeration and more like a marker of how far computational tools have come, and how audacious those early efforts truly were [2]. Fast forward to 2025. As I use the latest AI/ML tools available to a computational medicinal chemist, the value of knowledge earned the hard way is unmistakable. That long journey has paid off: two molecules designed through a careful blend of human insight and computational intelligence are currently in human trials.
Over the years, I have seen many researchers become obsessed with tools alone, only to fail and then blame the very technologies they relied upon. Witnessing this cycle repeatedly has shaped my perspective: I believe deeply in the potential of emerging technologies while respecting the immense and often unpredictable complexity of biological systems. There was always a ‘human in the loop’, and there always will be. Today, AI/ML has permeated nearly every discipline within the Chemical and Pharmaceutical Sciences [3]. Here, a broad, macroscopic view of how these emerging technologies are reshaping Chemical Sciences, and how human intelligence continues to remain central to their meaningful application, are presented (Chart 1).
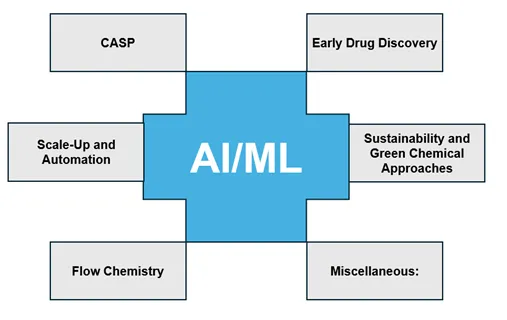
Chart 1. Overview of emerging applications of AI/ML in Chemical Sciences
CAMD and Drug Discovery in the Age of AI
Drug discovery, traditionally a slow and resource-intensive process, is being fundamentally reshaped by AI/ML. Modern CAMD tools now assist researchers at every early discovery stage, from identifying biological targets to designing molecules with higher probabilities of success [4]. By learning from vast chemical and biological databases, these tools can navigate expansive chemical spaces to generate and prioritise New Chemical Entities. Furthermore, they can flag potential safety or metabolism issues long before experimental work begins. This progress is driven by the collaboration between human intuition and machine intelligence. While AI rapidly explores chemical space beyond human capacity, chemists provide the judgment and biological understanding necessary to interpret results. This human-in-the-loop approach avoids blind reliance on algorithms and accounts for the inherent complexity of living systems. Ultimately, AI does not shorten discovery through brute force alone; it improves decision-making, reduces redundant experimentation, and increases the likelihood that promising lead molecules reach clinical stages faster [2].
Computer-Assisted Synthesis Planning (CASP): When Chemistry Meets AI
For a synthetic chemist, one of the most common challenges is scout for a synthetic route for the designed NCEs, which need to be synthesized quickly, and in sufficient purity, before any testing can begin. At this early stage, practicality rules. The goal is not perfection, but speed: fewer steps, easily available starting materials, and chemistry that is likely to work the first time. This is where AI-assisted CASP tools step in (Figure 1). Modern CASP and retrosynthesis tools can suggest multiple synthetic routes for a target molecule by breaking it down into simpler components. They act like highly experienced assistants, proposing feasible routes while leaving the final decision to the chemist.
How do these tools know what to suggest? Their ‘chemical intelligence’ comes from learning patterns in vast databases of known chemical reactions [5]. Powered by ML and modern computing, they recognise how molecules have been made in the past and apply that knowledge to new problems. Some tools rely on well-established reaction patterns, while others learn transformations directly from data. The next challenge is to predict forward synthesis, i.e., what will happen when chemicals are reacted with each other. Unlike retrosynthesis, forward synthesis usually give one major outcome. While AI tools can often predict the product, estimating the reaction yield remains difficult. Chemistry is influenced by subtle factors that are hard to capture fully in data. To overcome this, researchers are increasingly combining ML with traditional chemical intelligence. These hybrid approaches acknowledge an important truth that AI is powerful, but chemistry is still complex and human insight remains essential.
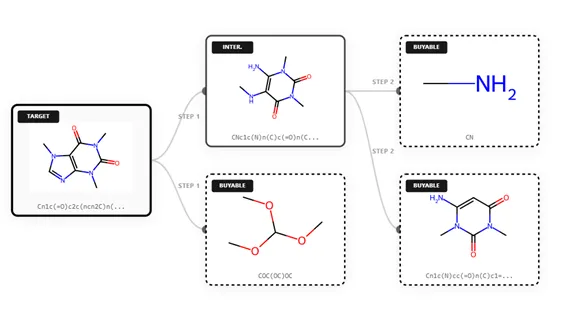
Figure 1. Representative retrosynthetic pathway (CASP) for caffeine (generated using
OCSR.ai proprietary tools)
AI/ML in Scale-Up, Safety, and Green Chemistry
The promising lead candidates have to be scaled-up safely and economically. Some modern AI tools can estimate whether a molecule is not scalable or not. They consider practical questions that chemists and engineers worry about every day including 1. Are the starting materials available in large quantities? 2. Is the cost reasonable? 3. Does the route avoid hazardous or unstable intermediates? and others [6]. More advanced approaches go a step further by acting as digital replicas of manufacturing plants. Trained in real process data, these AI models can simulate large-scale reactions and flag potential problems such as excessive heat buildup, poor mixing, or runaway reactions, before they happen. By identifying risks early, engineers can redesign processes virtually, reducing the chance of costly or hazardous failures during scale-up.
AI is also helping chemistry more environmentally friendly. One major focus is the replacement of toxic solvents with safer, more sustainable alternatives. Based on historic data, ML models can suggest greener substitutes that behave similarly in reactions but have a much lower environmental footprint. Another key concept in green chemistry is atom economy (how efficiently atoms from starting materials end up in the final product rather than becoming waste). Traditionally, calculating such metrics across many possible synthetic routes was slow and tedious. Today, AI-enabled tools can automatically evaluate thousands of proposed routes in seconds, highlighting those that generate less waste and use materials more efficiently. AI, in addition to molecular design, is increasingly shaping how chemicals are made, safer, cleaner, and smarter, while keeping human judgment firmly at the center of decision-making.
Flow Chemistry and AI-Driven Process Optimisation
Flow chemistry, a futuristic approach to manufacturing, involve reactions happening continuously as chemicals flow through microreactors. It offers better control, improved safety, and is naturally suited for digital monitoring, making it an ideal partner for AI/ML [7]. Exploring all possible combinations of variables such as temperature, pressure, reaction time, and reactants ratio, affecting the outcome, can be assigned as AI-based optimisation task, e.g., Bayesian optimisation. Instead of changing one variable at a time, or testing every possible combination, it learns from each experiment and intelligently decides what to try next. In practice, this means that reactions which once required hundreds of trials to optimise can now reach peak performance in just a few dozen experiments.
Flow chemistry also enables a new concept titled ‘self-optimising reactors’, which continuously analyse the reaction as happens using real-time sensors, or online chromatography. ML models interpret this data instantly to estimate yield and purity. Based on this feedback, the system can automatically adjust conditions such as flow rate or temperature to keep the reaction within desired quality limits. This level of control is essential for the shift toward continuous manufacturing, where consistency, safety, and efficiency must be always maintained. Together, flow chemistry and AI represent a powerful shift in how chemical processes are developed, moving from trial-and-error toward systems that learn, adapt, and optimise in real time, with chemists guiding the strategy rather than running endless experiments.
Challenges, Limits, and Ethical Questions
Despite its enormous promise, AI is not yet a plug-and-play solution for chemistry or drug development. One of the biggest challenges is data quality. AI systems learn from published literature and patents, which overwhelmingly report successful experiments. Failed reactions are rarely documented, even though they are essential for understanding the true limits of chemical reactivity. As a result, AI models are often trained on an incomplete and biased picture of reality. When noisy or flawed data go in, unreliable predictions come out.
Another concern is reproducibility and transparency. Many AI studies do not share their underlying code or datasets, making results difficult to verify. Even more problematic, some of the most powerful AI models function as black boxes. The ‘Why’ behind so-called accurate predictions is little understood or not understood at all. Regulation adds another layer of complexity. Regulatory agencies are now actively engaging with AI, developing frameworks that distinguish between low risk uses (such as early discovery) and high-risk ones (such as decisions affecting product release or patient safety). Regulators expect that the AI systems must be validated, auditable, and free from hidden bias before they can be trusted in critical settings.
Looking Ahead: The Next Phase of AI in Chemistry
The future of AI in Chemical Sciences is moving beyond prediction toward generation and autonomy (Table 1S, Supplementary Information section). Instead of merely suggesting options, future systems may design entire synthetic routes and experimental protocols from scratch, much like large language models generate text today. In some laboratories, AI is already beginning to communicate directly with robotic systems, closing the loop between design and execution. Another exciting frontier lies at the intersection of quantum chemistry and ML. By combining first-principles physics with data-driven models, researchers are working toward highly accurate ‘digital twins’ of chemical reactions. Such models could drastically reduce the need for trial-and-error experimentation, particularly in areas like catalyst design, where tiny electronic effects have outsized consequences. Sustainability will also become a built-in objective rather than an afterthought. Future AI systems are likely to evaluate environmental impact alongside cost and performance, automatically discarding wasteful or toxic pathways before a chemist ever sees them.
Conclusions
AI/ML represents a fundamental shift in how Chemical Sciences are practiced. Modern AI systems can now perform tasks that once required years of human experience, from retrosynthetic planning to reaction prediction, often at unprecedented speed. Yet this transformation comes with responsibility. Gaps in data, lack of transparency, and evolving regulatory expectations must be addressed through collaboration between academia, industry, and government. Openness, reproducibility, and ethical deployment will determine whether AI becomes a trusted partner or a fragile shortcut. Ultimately, AI/ML is not intended to replace chemists. Its real value lies in removing repetitive optimisation and exploration, freeing scientists to focus on creativity, insight, and a deeper understanding of biology and chemistry.
References:
- Xu J, Hagler A. Chemoinformatics and Drug Discovery. Molecules. 2002;7(8):566–600. doi: 10.3390/70800566.
- Ferreira FJN, Carneiro AS. AI-Driven Drug Discovery: A Comprehensive Review. ACS Omega. 2025;10(23):23889-23903. doi: 10.1021/acsomega.5c00549.
- Karthikeyan A, Priyakumar UD. Artificial Intelligence: Machine Learning for Chemical Sciences. J Chem Sci (Bangalore). 2022;134(1):2. doi: 10.1007/s12039-021-01995-2.
- Dhunnoo P. Drug Discovery in the Age of AI. https://medicalfuturist.com/drug-discovery-in-the-age-of-ai (Accessed January 25, 2026)
- Hammer AJS, Leonov AI, Bell NL, Cronin L. Chemputation and the Standardization of Chemical Informatics. JACS Au. 2021;1(10):1572-1587. doi: 10.1021/jacsau. 1c00303.
- Wightwick S. How AI is quietly changing drug manufacturability. https://www.drugtargetreview.com/article/181304/how-ai-is-quietly-changing-drug-manufacturability/ (Accessed on January 25, 2026)
- Alfano AI, García-Lacuna J, Griffiths OM, Ley SV, Baumann M. Continuous Flow Synthesis Enabling Reaction Discovery. Chem Sci. 2024;15(13):4618-4630. doi: 10.1039/d3sc06808k.
Supplementary Information
Key Concepts/Terms
- Forward Synthesis. Prediction of product of a chemical reaction between a set of reagents under specific conditions.
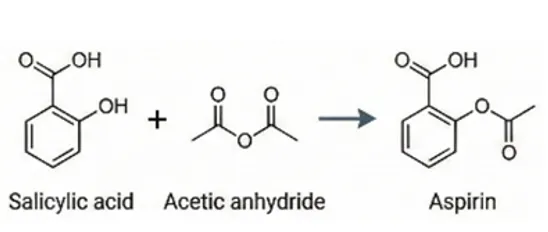
Figure 1S. Representative example of a forward synthesis
- Atom Economy. It is a well-accepted Green Chemistry tool. The expression of Atom Economy is –
It is expressed as % atom economy. Higher the value, greener is the process. Ideal reaction is where all the reactants are incorporated in the product with zero waste.
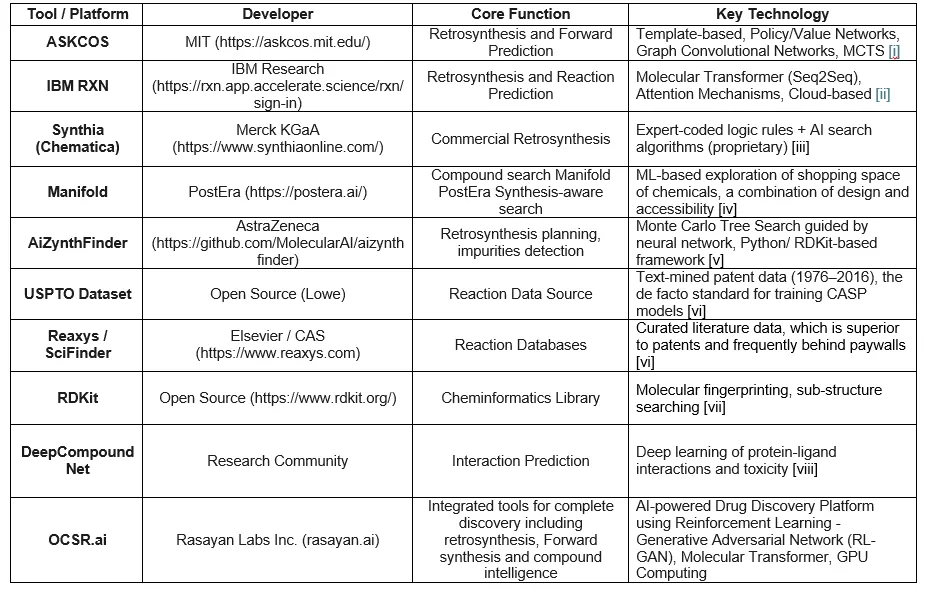
Table 1S. Summary of key AI tools, datasets, and platforms useful for a synthetic chemist
References
- Tu Z, Choure SJ, Fong MH, Roh J, Levin I, Yu K, Joung JF, Morgan N, Li SC, Sun X, Lin H, Murnin M, Liles JP, Struble TJ, Fortunato ME, Liu M, Green WH, Jensen KF, Coley CW. ASKCOS: Open-Source, Data-Driven Synthesis Planning. Acc Chem Res. 2025;58(11):1764-1775. doi: 10.1021/acs.accounts.5c00155.
- Schwaller P, Laino T, Gaudin T, Bolgar P, Hunter CA, Bekas C, Lee AA. Molecular Transformer: A Model for Uncertainty-Calibrated Chemical Reaction Prediction. ACS Cent Sci. 2019;5(9):1572-1583. doi: 10.1021/acscentsci.9b00576.
- Synthia™ Organic Retrosynthesis Software – Resources. https://www.sigmaaldrich.com/IN/en/technical-documents/technical-article/chemistry-and-synthesis/organic-reaction-toolbox/resources?utm_source=google&utm_medium=cpc&utm_campaign=15000381747&utm_content=129438265155&gad_source=1&gad_campaignid=15000381747&gbraid=0AAAAAD8kLQQA34PpnHVnCDS5FXbNGqtez&gclid=Cj0KCQiAm9fLBhCQARIsAJoNOcsx69KyJ7-laNKhMpOxtAF5-yCxliSrSqw814K6wVhbG2Mlr3BlhIAaAuymEALw_wcB (Accessed January 25, 2026)
- Medicinal Chemistry Powered by Machine Learning. https://postera.ai/ (Accessed January 25, 2026)
- Saigiridharan L, Hassen AK, Lai H, Torren-Peraire P, Engkvist O, Genheden S. AiZynthFinder 4.0: Developments Based on Learnings from 3 Years of Industrial Application. J Cheminform. 2024;16(1):57. doi: 10.1186/s13321-024-00860-x.
- Jiang Y, Yu Y, Kong M, Mei Y, Yuan L, Huang Z, Kuang K, Wang Z, Yao H, Zou J, Coley CW, Wei Y. Artificial Intelligence for Retrosynthesis Prediction. Engineering. 2023;25:32-50.
- Bento AP, Hersey A, Félix E, Landrum G, Gaulton A, Atkinson F, Bellis LJ, De Veij M, Leach AR. An open source chemical structure curation pipeline using RDKit. J Cheminform. 2020;12(1):51. doi: 10.1186/s13321-020-00456-1.
- Palhamkhani F, Alipour M, Dehnad A, Abbasi K, Razzaghi P, Ghasemi JB. DeepCompoundNet: Enhancing Compound-Protein Interaction Prediction with Multimodal Convolutional Neural Networks. J Biomol Struct Dyn. 2025;43(3):1414-1423. doi: 10.1080/07391102.2023.2291829.

Prashant S. Kharkar, a well-respected Drug Discovery Researcher with rich industrial experience, received his B. Pharm. degree in 1998. He obtained his PG and Ph.D. (Tech.) degrees Institute of Chemical Technology (ICT), Mumbai, India. Dr. Kharkar completed his postdoctoral research (2006–2009) on neurodegenerative diseases and disorders.

Akash Sirsat is pursuing M.Tech in Pharmaceutical Chemistry and Technology at the Institute of Chemical Technology (ICT), Mumbai. He completed his Bachelor of Pharmacy from Sinhgad Institute’s SKNCOP, Pune. His research interests include pharmaceutical chemistry, process development, and applications of AI/ML in pharmaceutical synthesis. He has qualified GPAT, GATE, NIPER, and GAT-B.








