Tuberculosis and in particular drug resistant tuberculosis is posing a major threat to the human population. Intensive research in related areas is in progress and many potential drugs were reported in literature. Many drugs are either heterocyclics or fused-heterocyclic systems and many other derivatives are in pipeline. Herein we have investigated a library of triazole based benzimidazole scaffold systems for their potential as anti-tuberculosis drugs by employing various computational pharmacokinetic and pharmacodynamics methods.
Introduction
Mycobacterium tuberculosis (Mtb)is one of the major infectious diseases posing a major threat to many developing countries including India. It has been a daunting task for researchers and scientists to design and develop drugs that possess the potential to inhibit the disease in a specific manner without exhibiting side effects in humans. Presently there are four drugs pyrazinamide, streptomycin, rifampinandisoniazid with therapeutic significance for the disease. Moreover, it is reported that improper usage of these drugs could lead to the development of drug-resistant strains1 of the causative bacterium and is therefore expected to be considered during the development process of design principles. Fused heterocyclics2 play critical roles in medicinal chemistry and are the backbone for many of the drugs available as anti-angiogenic, anti-oxidant, anti-microbial, anti-inflammatory, anti-cancer, etc.3Few examples of benzimidazole fused heterocycles along with their structures are given in table 1.
Table 1. Chemical structures of representative benzimidazole-based fused heterocyclics.
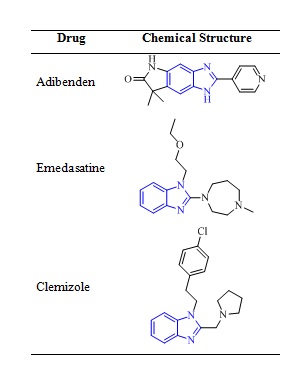
These benzimidazoles drugs are reported to inhibit the progression of tuberculosis by inactivating the MtbPrpR protein by binding to the key amino acids of MtbPrpR protein that plays significant role in methyl citrate cycle by activating many of the transcription factors.
With the emerging computer aided drug design, it possible to optimise the proposed drugs without being synthesized in lab, which is going to be both cost and time-effective. It also minimises the risk of drug toxicity and ensures to improve safety levels. For the recently emerged COVID-19 pandemic, many CADD based drugs were proposed and reported in the literature. Based on the above information along with several other associated advantages, herein we have investigated the design and inhibition potential of a library of potential drugs for tuberculosis and their ability to inactivate PrpR protein (PDB ID: 6D2S) of Mtb tuberculosis.4
Materials and methods
For the purpose of investigation we have used Glide docking to study the interactions between the drug and PrpR. Prime-MM/GBSA analysis5was employed for screening the drugs and molecular dynamic simulations were carried to validate the dynamic behavior of the inactivation process.
The structure of the proposed drugs was optimised and visualised using Biovia Discovery Studio Visualiser v16.1.0, (Dassault Systems Biovia software) before carrying the drug evaluation procedures.
A library of 20 triazole analogs (Figure 1) coupled with 1,2,5-tri substituted benzimidazole scaffolds were considered for evaluation. The initial structures of the analogs were drawn using ChemDraw Ultra 4.0 software.
The evaluation of drug likeliness was carried out following the principles of ADMET. All the docking calculations were performed using AutoDockVina algorithm following the conventional procedures.6
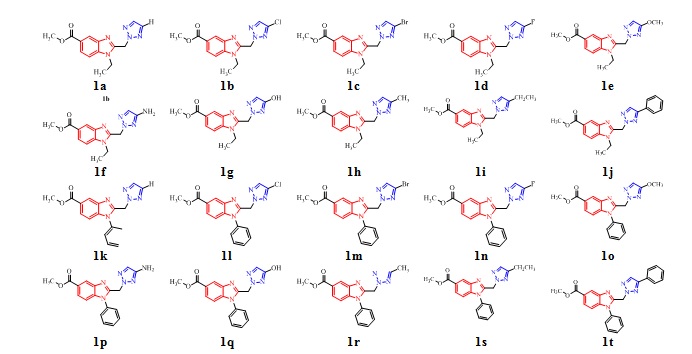
Figure 1. Chemical structures of the proposed library of triazole coupled 1,2,5-tri substituted benzimidazole scaffolds.
To predict the anti-tuberculosis activity, machine learning methods were employed to create models of proposed compounds, which have replaced the traditional slow and less reliable skin test.Herein we have adopted computational approaches to screen the active compounds against Mtb from a small proposed library.
Results and discussion
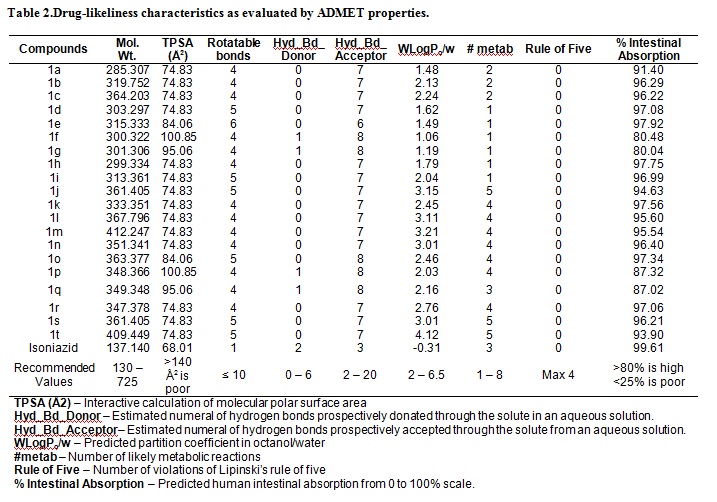
The evaluations made in this work are based on the reference and standard drug isoniazid. The ADMET calculations revealed satisfactory drug-likeliness of all the proposed compounds. The relevant data are compiled in table 2. It was revealed that all the compounds followed the Lipinski's rule of five and possesses enhanced absorption (> 80 %) by human intestine. Further it could be observed that compounds 1j – t, have enhanced distribution coefficient and hydrogen bond accepting capability which makes them as interesting targets for further investigations.
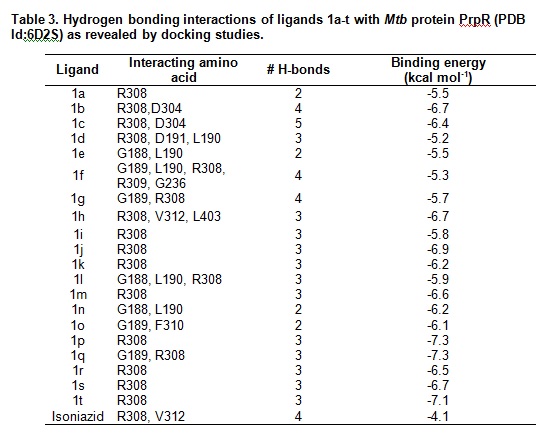
The docking calculations carried out to study the interaction of the compounds 1a – t with PrpR protein of Mtb and the relevant data are compiled in table 3. It was observed that all the compounds show very binding affinity towards the target PrpR of Mtbthan the standard isoniazid. The observed binding energy was in the range -4.1 to -7.3 kCal mol-1. Some of the amino acid residues involved in hydrogen bonding are G188, G189, L190, G236, R308, F310, V312 and L403. Compounds 1p, 1q and 1t exhibited relatively higher binding affinity towards the target than others and were considered for further investigations in this manuscript. The interactions of the above are depicted in figure 2.
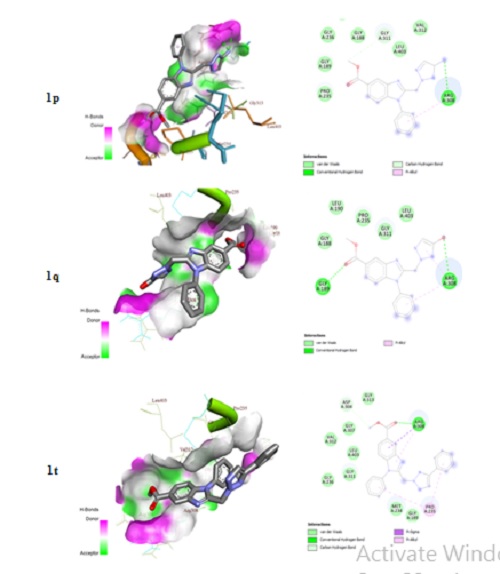
Figure 2. Binding interactions of 1p, 1q and 1t with PrpR protein (PDB Id: 6D2S) of Mtb as revealed from the best docked conformation.
In the case of 1p, a strong hydrogen bond was formed between R308 and the amine group of triazole moiety. Also van der Waals interaction was observed between G311 and the ester group of the 1p along with pi-alkyl interaction between the phenyl ring of 1p and R308. While in the case of 1q, hydrogen bond interaction was observed between triazole’s hydroxyl group of R308 and the ester group of G189 along with pi-alkyl interaction as observed in the above case of 1p. With 1t, the hydrogen bonding interaction was observed similar to that of 1p and 1q. Pi-alkyl interaction and van der Waals interaction was observed respectively between P235 & R308 and D304. The above results revealed the influence of lipophilic factors contributed by the strong electron donating groups and the heterocyclic rings.
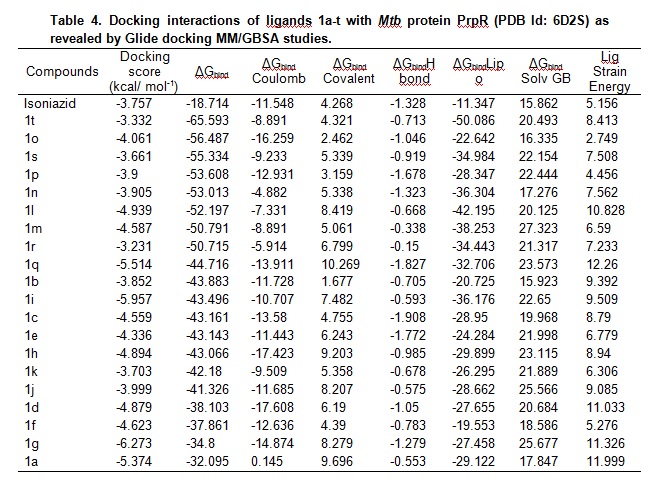
Further a detailed docking analysis was carried out using Glide7 followed byMM/GBSA and the corresponding results are given in table 4. It could be observed that compounds 1g, 1i and 1q have higher affinity to the active site of the target PrpR of Mtb than isoniazid and the corresponding binding energies are -6.273, -5.957 and -5.514 kcal mol-1 respectively as revealed by the docking scores. It is possible to calculate the free energies of binding of small molecules to biomacromolecules with high accuracy using MM/GBSA method. Though it could be seen in table 4 that many compounds have much lower free energy of binding than that top 3 scored compounds, the contributions from other factors could be responsible for the observed poor relative scores. Based on the binding free energies of -56.487, -55.334, -53.608 and -53.013 kcal mol-1 respectively for 1o, 1s, 1p and 1n, in comparison withresults of autodock, it can be concluded that compounds 1p, 1q and 1tcould be potential candidates for Mtb to inhibit or regulate transcription factors.
Further evaluations on the absorption, distribution, metabolism, excretion and toxicity studies were carried out with 1p, 1q and 1t, followed by molecular dynamic simulations to ascertain our above conclusions. Molecular dynamic simulations were carried out for the above hit molecules 1p, 1q and 1t in a time scale of 20 ns. The procedure was ascertained by employing PrpR-isoniazid complex as a reference for the analysis. The trajectories for 1p, 1q, 1t and standard isoniazid complexes with PrpR are shown in figure 3.
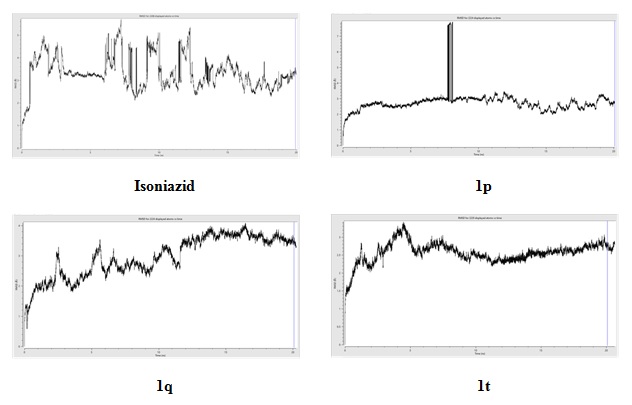
Figure 3. RMSD plot depicting the trajectories of 1p, 1q, 1t and Isoniazid in a time scale of 2 ns.
It is to be noted that at the end of 2 ns the hit compounds attained a variation of 2.8 Å, 3.4 Å and 2.8 Å respectively for 1p, 1q and 1t.
The minimum inhibitory concentrations8 of all 1p, 1q and 1t were evaluated computationally through machine learning web server mycoCSM9,10 and the corresponding values are given in table 5 and the results were comparable to that of experimentally calculated values.
Table 5. Predicted anti-tuberculosis activity by mycoCSM.
| Compounds | MIC(µM) |
| Isoniazid | 0.033608 |
| 1P | 0.008826 |
| 1q | 0.009123 |
| 1T | 0.008197 |
Conclusion
The pharmacodynamics and pharmacokinetics of triazole moiety with benzimidazole derivatives benzimidazole scaffolds, revealed their potential as anti-tuberculosis drug. All the results were comparable or on the better side of the control and standard drug Isoniazid. All the simulated parameters are in good agreement with the experimentally reported values.
References
[1] M. A. Espinal, Tuberculosis 2003,83 (1-3), 44.
[2] S. Rajasekhar, B. Maiti, M.M. Balamurali, K. Chanda, Curr. Org. Synth. 2017, 14, 40.
[3] A. Bistrovic, L. Krstulovic, A. Harej, P. Grbcic, M. Sedic, S. Kostrun, S. K. Pavelic, M. Bajic, S. Raic-Malic, Eur. J. Med. Chem. 2018,143, 1616.
[4] S. Tang, N. D. Hicks, Y. S. Cheng, A. Silva, S. M. Fortune, J. C. Sacchettini, Nucleic Acids Res. 2019, 47, 9934.
[5] G.M. Morris, R. Huey, W. Lindstrom, M.F. Sanner, R.K. Belew, D.S. Goodsell, A.J. Olson, J. Comput. Chem. 2009, 30, 2785.
[6] S. Genheden, U. Ryde, Expert Opin. Drug Discov. 2015, 10, 449.
[7] S. De Vita, G. Lauro, D Ruggiero, S. Terracciano, R. Riccio, G. Bifulco, J. Chem. Inf. Model. 2019, 59, 4678.
[8] D.E. Pires, D.B. Ascher, J. Chem. Inf. Model. 2020, 60, 3450.
[9] S. Jamal, S. Goyal, A. Shanker, A. Grover, Sci. Rep. 2017, 7, 872.
[10] M. Liu, Y. Wu, Y. Chen, J. Sun, Z. Zhao, X.W. Chen, M.E. Matheny, H. Xu, J. Am. Med. Inf. Assoc. 2012, 19, e28–35.

Soumyadip Das received a BSc degree from University of Calcutta, India, in 2017 and a MSc degree from Vellore Institute of Technology, Vellore, India, in 2019. He is currently pursuing his PhD degree from Vellore Institute of Technology, Vellore, India, under the supervision of Dr. Kaushik Chanda. His research interest includes diversity-oriented synthesis, anticancer drug design, and nanocatalysis.

Balamurali MM, was awarded the Master’s degree in Chemistry, by Madurai Kamaraj University, Tamil Nadu, India in 1999. He persuaded his doctoral research in Photophysical Chemistry at Indian Institute of Technology Kanpur, under the supervision of Prof. Sneh Kumar Dogra. In 2005, he was awarded the CSIR research associate fellowship and continued his postdoctoral research in Prof. Raghavan Varadarajan’s group at Indian Institute of Science Bangalore.

Kaushik Chanda, obtained his MSc in Organic Chemistry from Gauhati University, Assam India in 2001. Subsequently worked as a Senior Research Fellow in ICAR-NATP funded project in St Anthonys College, Shillong, India from 2002-2005. In 2006, he moved to Taiwan for pursuing PhD in Applied Chemistry from National Chiao Tung University under the guidance of Prof Chung Ming Sun on a topic of Combinatorial Chemistry.













