Metabolic Stability
A Critical Aspect in Drug Discovery
Rajesh Sonti, Assistant Professor, National Institute of Pharmaceutical Education and Research (NIPER)
Venkata Rao Kaki, Assistant Professor, National Institute of Pharmaceutical Education and Research (NIPER)
Siva Nageswara Rao Gajula, Pursuing PhD, National Institute of Pharmaceutical Education and Research (NIPER)
Nadimpalli Nimisha, Nadimpalli Nimisha pursued M.S. (Pharm.) in department of pharmaceutical Analysis,NIPER-Hyderabad.
Megha Sajakumar Pillai, Pursuing PhD, National Institute of Pharmaceutical Education and Research (NIPER)
Understanding metabolic stability helps to assess pharmacokinetic parameters for determining drug dosingand frequency. Enhancement of the drug’s metabolic stability helps improve patient compliance concerning dosing and dose frequency. Measuring metabolic study by microsomal t1/2 approach combined withthe automated LC-MS/MS and sample pooling strategies delivers reliable data with high-throughput screening.
The liability of a drug substance to the metabolism is referred to as metabolic stability[1]. The liver uses CYP-mediated metabolism to clear most orally administered drugs. Metabolic stability data is essential for evaluating and optimising the human therapeutic dose for the drug. In vitro systems obtained fromthe human liver, such as microsomes and hepatocytes, serve as dominant models to assess the metabolic stability of the drug [2-4]. Human liver microsomes offer numerous advantages, i.e., such as ease of availability and use for high-throughput screens but lack phase II metabolising enzymes, unlike hepatocytes[5, 6].Predicting in vivo pharmacokinetic data using in vitro drug metabolism data in preclinical investigations is of tremendous relevance. The in vitro metabolism data aids in selecting promising leads for further research. Drug clearance gives information on the dose required for maintaining steady-state plasma concentration and drug disposition[7, 8].
2. Significance of metabolic stability in drug design
The drug discovery and development processrelies on pharmacokinetics and metabolism studies. Pharmacokinetics and metabolism data from animal studies offer trustworthy physiologically relevant information [9]. In vitro studies are preferred as the in vivo studies are time-consuming and more expensive. In vitro drug metabolism studies with high-throughput screening have become more popular as evidence of a high link between in vitro and in vivo metabolic data has grown[10]. Drug discovery isstreamlined to be a more precise, fast, and iterative process. It is a continuous and iterative process to improve the structural scaffolds of drugs by medicinal chemists for identifying the ideal molecule. Maximum output is achieved by quickly generating and interpreting data from the metabolism studies for the designed drug. Knowledge of metabolic soft spots in a drug structure is crucial at the lead identification stage. This insight enables them to make structural alterations in the lead optimisation process to improve metabolomic stability. Further, it will be convenient to gather information on the impact of various structural modifications on metabolic stability during the tuning of pharmaceutical activity[11].
3. Approaches to improve the metabolic stability
The key to optimum drug design is to identify the best combination of several properties. Improvement of metabolic stability results in the enhanced plasma half-life of the drug substance. Moreover, it is essential to assess critical parameters such as elimination or reducing first-pass metabolism, enzyme induction, enzyme inhibition, and reactive or toxic metabolites [12]. Figure 1 illustrates the strategies to enhance the metabolic stability of a drug substance. In general, increasing the hydrophilicity or polarity by incorporating isosteric groups and blocking or replacing metabolic soft spots with other stable groups of the drug substances enhances metabolic stability[13]. Decreasing the drug’s lipophilicity reduces favourable binding contact with the CYP enzymes due to reduced hydrophobic interactions, decreasing the drug’s metabolism. In addition, the polarity of the molecule can also be increased by incorporating the heteroatom in the benzene ring, e.g., benzene to more polar pyridine.
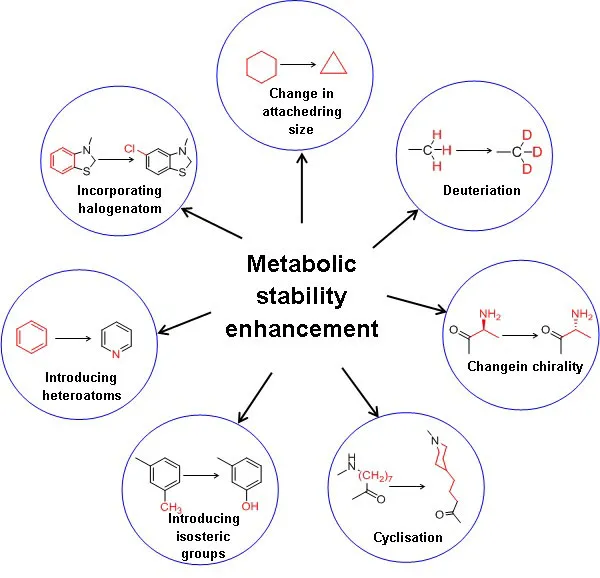
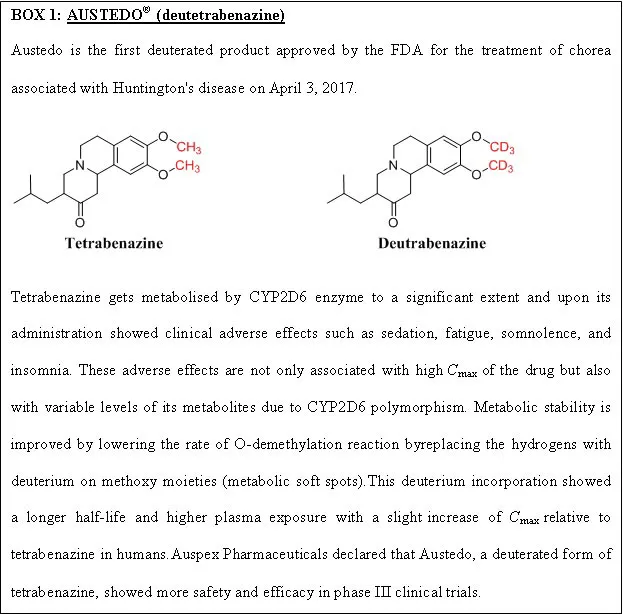
Deuterium incorporation at the potential metabolic soft spot is a novel strategy for enhancing metabolic stability with the same biological activity as the parent drug. The addition of deuterium to the molecule also affects the drug’s metabolic liabilities. The carbon deuterium bond is shorter on the order of 0.005 and stronger relative to the carbon-hydrogen bond due to the anharmonicity single bond vibrations. Moreover, deuterium has a lower vibrational frequency than hydrogen and takes more energy to break the carbon-deuterium bond. A kinetic deuterium isotope effect (KDIE) on the metabolic CLint is critical for applying a deuteration strategy to alter the drug pharmacokinetics. Wiberg calculated the theoretical maximum deuterium isotope effects (kH/kD) as 6.8 from the vibrational frequencies, and Bigeleisen formulated the theoretical base for such calculations[14]. When a drug has more than one metabolic oxidative soft spot, it is advised that all hydrogens in those locations be deuterated first, followed by a progressive reversion to hydrogen until the favourable outcome is lost by deuterium[15]. The extensive studies carried out for a drug in preclinical and clinical trials make the testing trials of the deuterated form faster and cost-effective. Deuterated drugs are metabolised at a slower rate and are less toxic due to fewer toxic metabolites formed and minimal drug-drug interactions. Since the first USFDA approval of deutetrabenazine (Box 1), many pharmaceutical companies have focused on deuterated drugs. Cyclisation, decreasing the size of the ring and chiral switching are the other strategies for enhancing metabolic stability. In comparison, the change in the chirality from S to R increased the half-life from 14 to 84 min[16]. The metabolism is reduced when labile groups are inserted into the cyclic structure. N-demethylation metabolic reaction can be avoided by inserting methyl groups attached to nitrogen into a cyclic structure[16].
4. Current progresses in analytical techniques for high-throughput metabolic stability assessment
The hyphenation of the chromatographic separation with the mass spectrometry (LC-MS) results in a better detection limit. Significant advancements in ionisation techniques and analysers in liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) and the simplicity of automation resulted inthe more sensitive and faster assessment of metabolic stability. Using sample pooling strategies (Cassette analysis) and a sensitive ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) offers high throughput screens. Different samples were pooled together in cassette analysis, decreasing the analysis time by a factor proportional to the number of pooled samples. However, this method has significant pitfalls, such as compound to compound interference, unpredictability in the analysis of weak ionisable drugs due to dilution from pooling, and coelution of isomers. Multiplex analysis, or the simultaneous measurement of multiple compounds in a single sample, became possible with the recent advancements in high-resolution mass spectrometers (HRMS). The multiplexed arrangement of the LC system allows the elution from multiple columns followed by MS detection. As with a single column LC system, this method minimises the injection-to-injection time while maintaining chromatographic integrity[17]. Nagele et al. performed a high-throughput metabolic stability study using the column switching technique by HRMS[18]. An ultra-fast method using exact mass TOF-MS utilising both ballistic gradient LC technique and sample pooling strategies has established an outstanding potential for measuring metabolic stability while addressing the drawbacks of the previous methods[19]. Acoustic droplet ejection technology delivers rapid, precise, and accurate nanoliter volumes of liquids on a hertz time scale. Acoustic energy creates an amplified standing wave in the sample well-containing fluid and ejects droplets. A piezoelectric transducer generates a focused ultrasonic pulse with an input waveform at 10 MHz centre frequency at each well of the source microplate. This ultrasonic pulse propagates through the coupling fluid into the walls and contents of the source microplate and reflects at the interfaces. Upon reflection, the pulse returns to the piezoelectric transducer for real-time processing to examine the microplate. Later, the application of a burst pulse results in thetransfer of droplets acoustically.The open port interface permits the capture and transfer of the acoustically ejected droplets to the electrospray source of the mass spectrometer. This open port interface alignsvertically with a co-axial tube arrangement that opens only on one end to receive the droplets. The inner tube connects to the capillary of the ESI source through a transfer line. A low-pressure liquid pump supplies carrier solvent to the outer tube to carry the ejected droplets. A nebuliser gas provides the aspirating pull for drawing the solvent from the outer tube into the inner tube.
5. Causes for the discrepancy observed between the calculated CLint from in vitro metabolising systems and the in vivo CLint
Many assumptions are made for evaluating in vivo intrinsic clearance from in vitro intrinsic clearance leading topotential drawbacks, and that may be erraticin certain circumstances.
(1) Metabolism in extrahepatic tissues: The anticipated in vivo CLint from the in vitro CLint will be underpredicted if the drug’s extrahepatic metabolism is significant. While the liver is an important site for drug metabolism, drugs can also undergo metabolism in other tissues. For instance, cyclosporine gets metabolised in the small intestine by CYP450 enzymes before reaching into the systemic circulation[20].
(2)Active transport through the sinusoidal membrane: If the drug is being actively transported via the sinusoidal membrane, the unbound concentration of drug in the hepatocytes compared to blood will be higher in the case of influx and lower in the case of efflux resulting in an over estimation or under estimation of in vivo CLint, respectively.
(3)Equilibrium between blood and hepatocytes: In the case of in vitro systems, metabolism of a drug occurs in hepatocytes, whereas in the in vivo condition, various models assume that there exists a rapid equilibrium of drug concentrations between blood and hepatocytes. Assume that the CLint in the hepatocytes exceeds the efflux clearance from hepatocytes to blood. The rate-limiting process for in vivo CLint is the influx of drugs from the blood to the hepatocytes, resulting in lower in vivo CLint than in vitro CLint
(iv)Inter-individual variability: Inter-individual variability arises across in vitro and in vivo metabolising systems. It was reported that the expression of CYP enzymes is different between the sexes. Hence, the pooling of microsomes or hepatocytes can minimise the problems related to inter-individual variability in thein vitro studies.
6. Importance of the integration of metabolic stability data with other high-throughput screening data
Choosing a screen within a drug discovery project should be based on a rational assessment of the project requirements. Furthermore, the throughput improvement is worthless without rigorous and continuous validation of the complete screening process[21]. Therefore, a combination of high-throughput screens, ADME screens, and metabolic stability studies are vitalin the drug discovery process. It is also conceivable that the self-inhibition of one or more drug-metabolising enzymes improves in vitro CLint. In general, the in vivo dose of a drug is more than that used for the in vitro studies. This higher dose in preclinical studies can lead to saturation of drug metabolising enzymes resulting in zero-order metabolism and enzyme inhibition. As a result, inhibition screening is recommended to combine with in vitro metabolic stability studies[11].
Conclusion
One of the crucial aspects ofthe drug research and development process is evaluating the drug’s metabolic characteristics. The pharmacokinetics of a drug candidate can be optimised in the drug discovery stage with the knowledge of metabolic stability. In the preclinical stage, all the major pharmaceutical companies have established in vitro methods to predict the drug metabolic properties. It was evident that the liver microsomes offer in vitro- in vivo extrapolation with high-throughput analysis. Furthermore, rapid and advanced improvements in the LC-MS/MS combined with unique analytical methodologies result in more accurate and fast results. In summary, integrating metabolic stability studies into the drug discovery process allows pharmaceutical industries to identify new potent and therapeutically active chemical entities at a lower cost and less time.
References
1. Masimirembwa, C.M., U. Bredberg, and T.B. Andersson, Metabolic stability for drug discovery and development. Clinical pharmacokinetics, 2003. 42(6): p. 515-528.
2. Hoener, B.A., Predicting the hepatic clearance of xenobiotics in humans from in vitro data. Biopharmaceutics & drug disposition, 1994. 15(4): p. 295-304.
3. Schneider, G., P. Coassolo, and T. Lavé, Combining in vitro and in vivo pharmacokinetic data for prediction of hepatic drug clearance in humans by artificial neural networks and multivariate statistical techniques. Journal of medicinal chemistry, 1999. 42(25): p. 5072-5076.
4. Lau, Y.Y., et al., Development of a novel in vitro model to predict hepatic clearance using fresh, cryopreserved, and sandwich-cultured hepatocytes. Drug metabolism and disposition, 2002. 30(12): p. 1446-1454.
5. McGinnity, D.F., et al., Evaluation of fresh and cryopreserved hepatocytes as in vitro drug metabolism tools for the prediction of metabolic clearance. Drug Metabolism and Disposition, 2004. 32(11): p. 1247-1253.
6. Gomez-Lechon, M., et al., Human hepatocytes as a tool for studying toxicity and drug metabolism. Current drug metabolism, 2003. 4(4): p. 292-312.
7. Bardal, S.K., J.E. Waechter, and D.S. Martin, Chapter 2—Pharmacokinetics. Applied pharmacology. Philadelphia: Content Repository Only, 2011: p. 17-34.
8. Chiou, W.L., The physiological significance of total body clearance in pharmacokinetic studies. Journal of Clinical Pharmacy and Therapeutics, 1982. 7(1): p. 25-30.
9. Zhang, D., et al., Preclinical experimental models of drug metabolism and disposition in drug discovery and development. Acta Pharmaceutica Sinica B, 2012. 2(6): p. 549-561.
10. Baranczewski, P., et al., Introduction to in vitro estimation of metabolic stability and drug interactions of new chemical entities in drug discovery and development. Pharmacological reports, 2006. 58(4): p. 453.
11. Thompson, T.N., Early ADME in support of drug discovery: the role of metabolic stability studies. Current drug metabolism, 2000. 1(3): p. 215-241.
12. Kumar, G.N. and S. Surapaneni, Role of drug metabolism in drug discovery and development. Medicinal research reviews, 2001. 21(5): p. 397-411.
13. Humphrey, M. and D. Smith, Role of metabolism and pharmacokinetic studies in the discovery of new drugs—present and future perspectives. Xenobiotica, 1992. 22(7): p. 743-755.
14. Bigeleisen, J., M. Wolfsberg in I. Prigogine. Advances in Chemical Physics, Interscience, New York, 1958. 1: p. 15-76.
15. Cargnin, S., M. Serafini, and T. Pirali, A primer of deuterium in drug design. 2019, Future Science.
16. MacKenzie, A.R., et al., Structure− Activity Relationships of 1-Alkyl-5-(3, 4-dichlorophenyl)-5-{2-[(3-substituted)-1-azetidinyl] ethyl}-2-piperidones. 1. Selective Antagonists of the Neurokinin-2 Receptor. Journal of medicinal chemistry, 2002. 45(24): p. 5365-5377.
17. Zhang, J., et al., A high‐speed liquid chromatography/tandem mass spectrometry platform using multiplexed multiple‐injection chromatography controlled by single software and its application in discovery ADME screening. Rapid Communications in Mass Spectrometry, 2013. 27(7): p. 731-737.
18. Nägele, E. and A.S. Fandino, Simultaneous determination of metabolic stability and identification of buspirone metabolites using multiple column fast liquid chromatography time-of-flight mass spectrometry. Journal of Chromatography A, 2007. 1156(1-2): p. 196-200.
19. Manna, J.D., S.J. Richardson, and M.F. Moghaddam, Implementation of a novel ultra fast metabolic stability analysis method using exact mass TOF-MS. Bioanalysis, 2017. 9(4): p. 359-368.
20. Hebert, M.F., et al., Bioavailability of cyclosporine with concomitant rifampin administration is markedly less than predicted by hepatic enzyme induction. Clinical Pharmacology & Therapeutics, 1992. 52(5): p. 453-457.
21. Eddershaw, P.J. and M. Dickins, Advances in in vitro drug metabolism screening. Pharmaceutical science & technology today, 1999. 2(1): p. 13-19.

Rajesh Sonti is working as an Assistant Professor since Feb 2020 in Department of Pharmaceutical Analysis, National Institute of Pharmaceutical Education and Research (NIPER)-Hyderabad. He completed PhD from IISc, Bangalore and later worked as a post doc in University of Basel and EPFL-Lausanne for 6.5 years.

Venkata Rao Kaki is working as an Assistant Professor in Department of Chemical Sciences (medicinal chemistry/natural products),NIPER-Hyderabad. He completed PhD from Panjab university, Chandigarh. He has 4 years industrial and 8 years of academic experience.

Siva Nageswara Rao Gajulais pursuing PhD in Department of Pharmaceutical Analysis, NIPER-Hyderabad. He is working in the scientific areas:Drug Metabolism and Pharmacokinetics (DMPK), biomarkers and drug-drug interactions. He did M.S. (Pharm.) in department of pharmaceutical Analysisfrom NIPER-Ahmedabad.

Pursued M.S. (Pharm.), National Institute of Pharmaceutical Education and Research (NIPER)

Megha Sajakumar Pillai pursued M.S. (Pharm.) in department of pharmaceutical Analysis, NIPER-Hyderabad. She is currently pursuing PhD in NIPER-Ahmedabad.






