Redefining Therapeutics
Immune modulatory vaccines as pipeline-ready combination amplifiers
Mads Hald Andersen, DMSc, DScTech, PhD, Director, Professor National Center for Cancer Immune Therapy, Copenhagen University Hospital Herlev
Immune modulatory vaccines (IMVs) are a novel immunotherapeutic modality for cancer treatment. IMVs expand endogenous T cells that recognise tumor microenvironment antigens (TMAs). Rather than blocking a single pathway, IMVs can eliminate or reprogram immune suppressive cells. IMVs may enhance the effect of anti-cancer therapy, including checkpoint inhibitors, vaccines, cellular therapies, chemo- and radiotherapy. IMVs may additionally enhance antiviral immunity and treat fibrosis.
Introduction
Immune modulatory vaccines (IMVs)
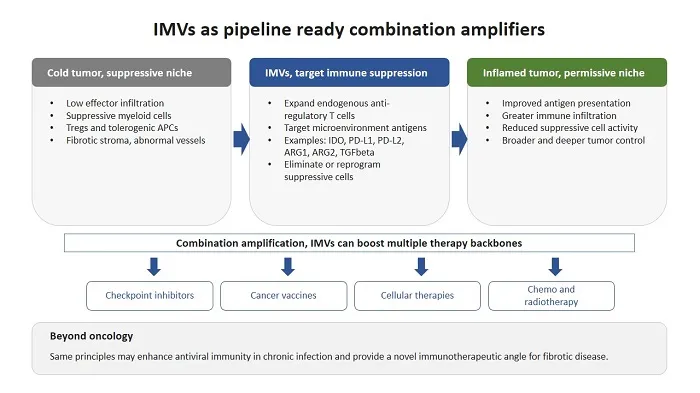
Cancer immunotherapy has proven that removing inhibitory signals can generate durable tumor control, yet it has also clarified the core limitation. Many treatments do not fail because effector cells cannot recognise cancer, but they fail because the local ecosystem enforces immune suppression and immune exclusion. The tumor microenvironment contains regulatory T cells, suppressive myeloid populations, tolerogenic antigen presenting cells, abnormal vasculature, and fibrotic stroma, coordinated by inhibitory cytokines and metabolic constraints that collectively restrict effective immunity (1).
IMVs are an emerging therapeutic class designed to target this bottleneck (2). Instead of vaccinating against tumor associated antigens or neoantigens, IMVs vaccinate against tumor microenvironment antigens (TMAs) (3). These are host-derived proteins expressed by immune regulatory cells and include metabolic enzymes, immune checkpoints, and regulatory cytokines, for example IDO1, PD-L1, PD-L2, Arginase 1 (ARG1), Arginase 2 (ARG2), and TGFβ. These molecules are expressed not only by malignant cells, but also by regulatory and stromal cell populations in the tumor microenvironment. In principle, IMVs can be implemented using any vaccine platform, allowing pharmaceutical developers to choose the format that best fits their capabilities and pipeline strategy. To date, most clinical experience and development has been generated with peptide-based IMVs formulated with Montanide as adjuvant, while RNA based IMVs are also already in active development.
For the pharmaceutical community, the central message is that IMVs can be viewed as pipeline ready combination amplifiers, a platform modality that can be layered onto existing therapies with minimal added toxicity. By reshaping suppressive niches, IMVs can deepen responses, extend durability, and broaden the responder population, including patients with PD-L1-low or immune excluded tumors, where checkpoint blockade alone has limited effect (4-6).
Primary mechanisms of action.
IMVs are defined as vaccines designed to induce antigen-specific immune responses against TMAs, with the primary aim of eliminating or reprogramming immunosuppressive cell populations rather than only targeting malignant cells. IMVs activate a naturally occurring component of immune regulation, self-reactive T cells termed anti-regulatory T cells (7). Anti-regulatory T cells recognise HLA-restricted peptides derived from TMAs and can counteract immune suppression by killing suppressive cells and by reprogramming suppressive compartments through inflammatory cytokines. They exist at low frequencies in healthy individuals and can expand in response to inflammatory stimuli, functioning as physiological sentinels that help prevent unchecked accumulation of suppressive cell states.
IMVs activate both CD8 and CD4 T cells. CD8 T cells predominantly mediate direct cytotoxic killing of tumor microenvironment antigen-expressing suppressive cells. CD4 T cells predominantly secrete pro-inflammatory cytokines that reprogram regulatory and stromal populations toward immune permissive phenotypes. Through these combined cytolytic and modulatory mechanisms, IMVs remodel suppressive cellular networks, enhance antigen presentation, promote immune infiltration, and amplify endogenous tumor-specific immunity.
A major difference between IMVs and other modalities targeting immune suppression is that IMVs operate at the level of cellular sources of suppression rather than single molecular pathways. Eliminating or reprogramming a suppressive myeloid population removes not only one enzyme or one ligand, but also coupled outputs such as IL10, TGFβ, reactive oxygen species, and adenosine. Targeting cancer-associated fibroblasts can reduce stromal rigidity and chemokine mediated immune exclusion (8). This ecosystem-level impact is a key mechanistic difference from single target antibodies and inhibitors, which typically block one pathway while leaving the cellular producers of suppression intact. The failure of Phase III trials with small molecule IDO1 inhibitors illustrates that enzymatic blockade alone may be insufficient to improve clinical outcomes (9). An IDO1-based IMV differs fundamentally from an IDO1 small molecule inhibitor, as it induces adaptive cellular immunity that targets IDO1-expressing suppressive cells and induces local inflammation. For pharma pipelines, this supports IMVs as a way to convert a pathway into an immune target, complementing inhibitor programs and opening opportunities to revisit targets that have disappointed as stand-alone pharmacologic inhibitors.
Use of IMVs in cancer
IMVs as monotherapy
The most clinically advanced IMV programs target two tumor microenvironment antigens, IDO1 and PD-L1. Early peptide vaccination studies against these targets showed that vaccination was well tolerated and consistently induced vaccine-specific T-cell responses (10-12). Across settings including non-small cell lung cancer, lymphoma, and multiple myeloma, IMVs demonstrated a favorable safety profile with early indications of activity. This low toxicity profile supports development not only in combination regimens, but also as off-the-shelf monotherapy in early disease settings, including neoadjuvant and adjuvant treatment, where therapeutic vaccination has already shown promise (2). Beyond IDO1 and PD-L1, next generation IMVs target ARG1 and TGFβ to address metabolic suppression and fibrotic exclusion, aiming to convert cold tumors into inflamed, immune permissive tissue and amplify multiple classes of therapies (8, 13).
Combination amplification with immune checkpoint inhibitors
The clearest clinical evidence that IMVs can function as combination amplifiers comes from IDO1 and PD-L1 targeting IMVs. In the Phase II MM1636 trial, a long IDO1-derived peptide (IO102) and a long PD-L1-derived peptide (IO103) were formulated with Montanide as adjuvant and administered in combination with nivolumab as first line therapy for metastatic melanoma.(14). With extended follow up, median progression free survival reached 25.5 months and median overall survival was not reached after almost four years, comparing favorably to matched historical controls receiving anti-PD1 alone (4).
Correlative data supported a mechanistic division of labor. Immune monitoring confirmed induction of vaccine-specific CD4 and CD8 T cells that trafficked into tumors and reprogrammed suppressive myeloid populations (14). Serum proteomic profiling identified a vaccine-specific immune signature characterised by early induction of CCL3, CCL4, and TNFα, not observed in a matched patient cohort receiving anti-PD1 monotherapy, indicating distinct systemic immune modulation contributed by the IMV (4).
A Phase III trial then evaluated IO102 and IO103 in combination with pembrolizumab in more than 400 patients with advanced melanoma (6). Importantly, the regimen was well tolerated, with adverse events largely limited to low grade injection site reactions. The combination achieved a median progression free survival of 19.4 months compared with 11.0 months for pembrolizumab alone, representing the longest median progression free survival yet reported in a Phase III metastatic melanoma trial. Although the prespecified threshold for statistical significance was narrowly missed in the overall population, hazard ratio 0.77, p = p=0.056, clinical benefit was observed across all predefined subgroups, except patients previously treated with PD-1 blockade. In PD-1 naive patients, median progression free survival reached about 25 months, consistent with the Phase II results in a comparable population, versus 11.0 months in the control arm.
Importantly, this Phase III study also highlights that IMVs may extend the reach of immunotherapy into tumors with low or absent PD-L1 expression and immune excluded phenotypes, populations traditionally resistant to checkpoint blockade. This reframes IMVs as add-on agents that can broaden the addressable population for checkpoint backbones and reduce reliance on PD-L1 enrichment for clinical activity. Given that vaccination can increase expression of multiple checkpoint receptors on activated T cells, additional checkpoint blockade, including CTLA-4 or LAG-3, may further enhance efficacy by strengthening priming and sustaining intratumoral effector function. Finally, safety supports long term administration, enabling adjuvant programs aimed at preventing recurrence. IMVs can be administered over extended periods with limited systemic toxicity, which is essential in potentially curative settings where tolerability determines uptake.
Combination amplification with cancer vaccines
Tumor-specific vaccines can expand T cell repertoires, but their clinical impact can be limited by suppressive niches and suboptimal antigen presentation within the tumor. This makes it an obvious strategy to combine conventional tumor antigen vaccines with an IMV component targeting TMAs, thereby coupling tumor directed priming with active relief of immune suppression. In animal models, adding an IMV increases the antitumor activity of tumor directed vaccination, consistent with the idea that removing suppression allows vaccine induced T cells to function in the tumor (15). Mechanistically, IMVs can remodel the microenvironment by reducing suppressive cell activity, improving antigen presentation, and facilitating immune infiltration, which together increase the likelihood of effective intratumoral killing and broader antigen spread.
Combination amplification with cellular therapies
Adoptive cellular therapies in solid tumors are limited by suppressive myeloid barriers, metabolic restriction, and fibrotic exclusion (16). IMVs that remodel myeloid and stromal compartments provide a rational route to improve trafficking and persistence of infused cells and to increase the fraction of patients, who can benefit from cellular approaches.
Combination amplification with chemo and radiotherapy
Chemo- and radiotherapy can drive antigen release and inflammation, yet they can also induce counter regulatory programs including induction of IDO1, PD-L1, and TGFβ (17). IMVs are suited to exploit immunogenic components while countering suppressive rebound. Translational analyses in pancreatic cancer link baseline TGFβ-specific immunity with improved outcomes when radiotherapy and checkpoint blockade are combined, supporting stromal and cytokine targeting as a partner strategy in cold tumors.
IMVs beyond oncology
Infectious diseases
The same regulatory pathways that blunt antitumor immunity in cancer are also induced in chronic and acute infections, where immune suppression constrains pathogen clearance and limits antiviral immune responses (18). In chronic viral infections such as HIV, HBV, and HCV, TMAs including IDO1, PD-L1, contribute to regulatory niches and T cell exhaustion. Similar, but more transient, induction of these pathways can occur in acute infection, potentially limiting early viral clearance.
Although no IMVs have yet been tested clinically in infectious diseases, anti-regulatory T cells have repeatedly been shown to increase T cell immunity toward viral antigens. In vitro experiments demonstrated that PD-L1-specific T cells enhanced proliferation of influenza-specific and Epstein Barr virus-specific T cells, and IDO1-specific T cells augmented Epstein Barr virus-specific responses by reacting to IDO1-positive regulatory cells and reducing resident regulatory T cells (18).
From a translation perspective, the most plausible early opportunities are combination strategies. In HIV, IMVs directed against IDO1 and PD-L1 could reduce exhaustion and synergize with latency reversing agents to help eliminate viral reservoirs. In HBV and HCV, where antigen presenting cells often induce tolerance with PD-L1, PD-L2, and secretion of IDO1, arginase, and TGFβ, an IMV could serve as an enabling layer to improve therapeutic vaccination or augment prophylactic vaccine responses in low-responder populations.
Fibrosis as an immunotherapeutic target
Fibrosis is not only structural, it also actively shapes immune regulation (19). Chronic viral hepatitis illustrates this link, where persistent inflammation drives TGFβ production and extracellular matrix deposition that can progress to cirrhosis. Preclinical data show that TGFβ-specific T cells can reduce fibrosis, normalise stromal architecture, and improve tissue function (20). A similar rationale may apply to tuberculosis granulomas, where TGFβ and ARG1 contribute to fibrosis and encapsulation.
Conclusions
IMVs represent a distinct and increasingly validated immunotherapeutic modality. They expand anti-regulatory T cells against TMAs, activate both CD4 and CD8 T cells, combine direct cytolysis with cytokine driven reprogramming, and dismantle suppressive networks by targeting the cellular sources of suppression rather than a single pathway.
Clinically, IMVs have reached an important proof-of-concept milestone. Therapeutic vaccination combined with anti-PD1 therapy can improve progression free survival in metastatic melanoma, with tolerability compatible with long-term use and the clearest signals in PD1-naive and PD-L1-low settings.
For pharma, IMVs are pipeline ready combination amplifiers. As add-on agents, they can broaden responder populations, deepen and prolong benefit, and support earlier line and perioperative regimens. Similar principles may also apply beyond oncology, where immune suppression and fibrotic remodeling limit outcomes in chronic infection and fibrotic disease.
References
1. Xu J, Ding L, Mei J, Hu Y, Kong X, Dai S, et al. Dual roles and therapeutic targeting of tumor-associated macrophages in tumor microenvironments. Signal Transduct Target Ther. 2025;10(1):268.
2. Strum S, Andersen MH, Svane IM, Siu LL, Weber JS. State-Of-The-Art Advancements on Cancer Vaccines and Biomarkers. Am Soc Clin Oncol Educ Book. 2024;44(3):e438592.
3. Andersen MH. Tumor microenvironment antigens. Semin Immunopathol. 2022.
4. Pedersen S, Byrdal M, Martinenaite E, Lorentzen CL, Kjeldsen JW, Thorsen SU, et al. Five-year clinical outcome and immune biomarkers of durable response from the MM1636 trial on IDO/PD-L1 vaccination and PD-1 blockade in first line metastatic melanoma. Nat Commun. 2025.
5. Andersen MH. Immune modulatory vaccines: the safe way to inflame the tumor microenvironment & guide anti-cancer immunotherapies. Immuno-oncology Insights. 2022;3(9):7.
6. Hassel JC, Arance A, Carlino MS, Ascierto PA, Sandhu SK, Puzanov I, et al. LBA53 - IO102-IO103 cancer vaccine plus pembrolizumab for first-line (1L) advanced melanoma: Primary phase III results (IOB-013/KN-D18). Annals of Oncology. 2025;36:S1712–S3.
7. Andersen MH. Anti-regulatory T cells. Semin Immunopathol. 2017;39(3):317-26.
8. Andersen MH. Novel immune modulatory vaccines targeting TGFbeta. Cell Mol Immunol. 2023;20(5):551-3.
9. Cicin I, Plimack ER, Gurney H, Leibowitz R, Alekseev BY, Parnis FX, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab for advanced urothelial carcinoma: results from the randomized phase III ECHO-303/KEYNOTE-698 study. BMC Cancer. 2024;23(Suppl 1):1256.
10. Klausen U, Gronne Dahlager Jorgensen N, Grauslund JH, Munir Ahmad S, Gang AO, Martinenaite E, et al. An immunogenic first-in-human immune modulatory vaccine with PD-L1 and PD-L2 peptides is feasible and shows early signs of efficacy in follicular lymphoma. Oncoimmunology. 2021;10(1):1975889.
11. Jorgensen NG, Klausen U, Grauslund JH, Helleberg C, Aagaard TG, Do TH, et al. Peptide Vaccination Against PD-L1 With IO103 a Novel Immune Modulatory Vaccine in Multiple Myeloma: A Phase I First-in-Human Trial. Front Immunol. 2020;11:595035.
12. Kjeldsen JW, Iversen TZ, Engell-Noerregaard L, Mellemgaard A, Andersen MH, Svane IM. Durable Clinical Responses and Long-Term Follow-Up of Stage III-IV Non-Small-Cell Lung Cancer (NSCLC) Patients Treated With IDO Peptide Vaccine in a Phase I Study-A Brief Research Report. Front Immunol. 2018;19:(9):2145-50.
13. Andersen MH. The targeting of tumor-associated macrophages by vaccination. Cell Stress. 2019;3(5):139-40.
14. Kjeldsen JW, Lorentzen CL, Martinenaite E, Ellebaek E, Donia M, Holmstroem RB, et al. A phase 1/2 trial of an immune-modulatory vaccine against IDO/PD-L1 in combination with nivolumab in metastatic melanoma. Nat Med. 2021;27(12):2212-23.
15. Nandre R, Verma V, Gaur P, Patil V, Yang X, Ramlaoui Z, et al. IDO Vaccine Ablates Immune-Suppressive Myeloid Populations and Enhances Antitumor Effects Independent of Tumor Cell IDO Status. Cancer Immunol Res. 2022;10(5):571-80.
16. Zugasti I, Espinosa-Aroca L, Fidyt K, Mulens-Arias V, Diaz-Beya M, Juan M, et al. CAR-T cell therapy for cancer: current challenges and future directions. Signal Transduct Target Ther. 2025;10(1):210.
17. Zhang Z, Liu X, Chen D, Yu J. Radiotherapy combined with immunotherapy: the dawn of cancer treatment. Signal Transduct Target Ther. 2022;7(1):258.
18. Andersen MH. Immune modulatory vaccines: time to move into infectious diseases. Lancet Microbe. 2023;4(1):e4-e5.
19. Zhao M, Wang L, Wang M, Zhou S, Lu Y, Cui H, et al. Targeting fibrosis, mechanisms and cilinical trials. Signal Transduct Target Ther. 2022;7(1):206.
20. Perez-Penco M, Weis-Banke SE, Schina A, Siersbaek MS, Hubbe ML, Jorgensen MA, et al. TGFβ-derived immune modulatory vaccine: targeting the immunosuppressive and fibrotic tumor microenvironment in a murine model of pancreatic cancer. Journal for ImmunoTherapy of Cancer. 2022;10(12).

Professor Mads Hald Andersen, DMSc, DScTech, PhD, is Director of the National Center for Cancer Immune Therapy, CCIT-DK, at Copenhagen University Hospital Herlev, Denmark. He discovered tumor microenvironment antigens and anti-regulatory T cells and defined their role in immune regulation. He initiated the immune modulatory vaccine concept and has authored more than 250 publications and 25 patents.








