
The incidence of Central Nervous System (CNS) illness is on the upswing and exacts a heavy human and economic toll. According to the World Health Organization, more than 120 million people worldwide suffer from depression, and the number is expected to rise. The occurrence of Alzheimer’s disease is slated to grow by more than 100 per cent in developed countries by 2040 and by 300 per cent in China, India, South Asia and Western Pacific countries over the same time period. In the United States, the National Mental Health Association estimates the annual cost of direct treatment of mental illnesses, the social costs of leaving it untreated and lost productivity at US$ 205 billion.
With these statistics as a backdrop, it is worth noting that hundreds of CNS therapies are in development, more than 300 in the United States alone, yet they lag behind in the development of therapies for non-CNS disorders in terms of the time needed to bring them to market. Data suggest that it takes 12.6 years on average for CNS agents to obtain regulatory approval, twice as long as cardiovascular agents. In addition, a mere 7 per cent of investigational CNS drugs that start in clinical development are eventually marketed as compared to 15 per cent for non-CNS candidates.
The reasons are many as to why CNS drugs are more difficult to develop successfully, namely the sheer complexity of the brain; the presence of CNS-mediated side effects such as nausea, dizziness and seizures; the need for agents to pass the blood-brain barrier; and the lack of validated biomarkers. Yet, with the implementation of targeted strategies, it may be possible to identify more quickly and with greater accuracy those therapeutics agents with the most promise, leading to better GO / NO-GO decisions.
This article focusses on selected techniques that can be used early on, following preclinical work, to launch Phase I studies that determine if compounds are entering the brain, and how this action may be impacted by dose ranging. These techniques are the routine Electroencephalogram (EEG), the Quantitative Electroencephalogram (QEEG) and Evoked Potential (EP). They can be used as tools for translating preclinical findings into first-in-human studies. They may also play a role in later phase studies when compounds shift from being studied in healthy volunteers to CNS patients. Additional translational techniques employed in Phase I programmes include cerebral spinal fluid sampling, brain imaging methodologies, and cognitive and behavioural assessments. These are beyond the scope of this paper.
 The Techniques
The Techniques
The EEG, QEEG and EP are non-invasive techniques with results recorded from electrodes attached to the surface of the scalp according to an internationally standardised arrangement, the so called 10-20 system (Figure 1). The promise for these methods is to be able to predict, early on in the clinical development of novel therapies that an antidepressant or analgesic will be safe and efficacious at various doses as compared to placebo. Moreover, the EEG or EP signature observed, including dose versus response relationships observed, in animals can be translated and validated against healthy human volunteers and in subjects with CNS disorders.
The EEG, QEEG and EP are well-established measures of electrical patterns in the brain, but their power as tools for translating preclinical findings into Phase I trials is just beginning to be applied. Depending upon the therapeutic area—Alzheimer’s disease, Schizophrenia, pain, Major Depression, Parkinson’s disease, or sleep disorders—all or some of the three techniques might prove useful in characterising compounds starting into Phase I clinical trials.
 Routine EEGs, for example, can be used for monitoring CNS toxicity and for detecting increased seizure likelihood. They provide a continuous measure of cortical function with excellent time resolution, measurable in milliseconds. Unlike relatively new functional imaging procedures, such as Single-Photon Emission Computed Tomography (SPECT), Positron Emission Tomography (PET), and functional MRI (fMRI), EEG has the advantage of being well tolerated, easy to administer, and relatively inexpensive.
Routine EEGs, for example, can be used for monitoring CNS toxicity and for detecting increased seizure likelihood. They provide a continuous measure of cortical function with excellent time resolution, measurable in milliseconds. Unlike relatively new functional imaging procedures, such as Single-Photon Emission Computed Tomography (SPECT), Positron Emission Tomography (PET), and functional MRI (fMRI), EEG has the advantage of being well tolerated, easy to administer, and relatively inexpensive.
Quantitative EEGs are more sophisticated as they can monitor the time course of CNS effects of compounds, detail typical profiles of drugs in proof of concept studies and monitor vigilance effects, which refers to the brain’s state of receptivity to external stimuli associated with alertness. Both types of EEGs can be used in conjunction with fMRI, which, while slower to react, in the range of seconds, offers better spatial resolution and could corroborate EEG findings. Together, they can offer a higher degree of confidence that there is a useful signal to go forward.
Evoked potential refers to an electrical potential recorded from a subject following presentation of an acoustic or visual stimulus, and can be a surrogate biomarker for cognitive information processing and for neurotransmitter systems such as serotonin. The ability to monitor serotoninergic function is valuable because of its putative role in the pathophysiology and in therapeutic interventions for CNS disorders. An EP method known as the Loudness Dependence Auditory Evoked Potential (LDAEP) can serve as an indicator of central serotonin function in animals and humans and may show promise as a predictor of selective serotonin reuptake inhibitor antidepressant treatment response. With the LDAEP method, various techniques, such as dipole source analysis and scalp topography analysis, may detect the effect of enhanced serotonin availability.
One component of EP, known as Mismatch Negativity (MMN) is particularly useful as a biomarker for schizophrenia. MMN is a response that a subject has a variation within a sequence of regular stimuli and is a reflection of sensory memory. For example, if there is repetitive auditory stimulation, such as “b-b-b-b-b”, interspersed with an occasional “r”, the subject elicits an MMN response. Furthermore, the MMN response grows in correlation with the number of repetitions of the standard stimulation. Subjects with chronic schizophrenia, however, tend to generate impaired MMN, indicating that they recognise the change within the sequence in a less robust manner than healthy volunteers and the impaired response may grow as the disease progresses.
Although, schizophrenia therapies in development can be tested in healthy volunteers, frequently the sensitivity to detect a relevant therapeutic surrogate for the drug effect expected in the disorder can be either enhanced or reduced based on the neurophysiological differences between patients and healthy volunteers. MMN response is a tool that can be used to explore the potential differences in treatment effect between patient populations and volunteers. If there is a normalisation response among the schizophrenic patients, this would be indicative of drug action, i.e. normalising the observed mismatch negativity. This electrophysiological endpoint might serve as a surrogate marker, convergent with the cognitive and information deficit symptoms observed in the disorder. Even if surrogate marker validation is preliminary, the observed effect demonstrates that the compound is crossing the blood-brain barrier and is showing evidence of efficacy.
Using the techniques
A number of studies have been conducted that demonstrate the value of using the described methods to translate preclinical findings into Phase I trials. This section is not intended as a review of all of the studies in this area, but rather, it is meant to highlight a few examples of how the electrophysiological techniques have been applied.
Parker et al. (2001) report using the quantitative EEG approach in an animal study to define the pharmacokinetic / pharmacodynamic (PK / PD) profile of two investigational antipsychotic compounds, known as S18327 and S16924. Clozapine, also an anti-psychotic, served as an active control. Qualitative EEG changes in this animal model were similar for clozapine, but different for the investigational compounds potentially indicating a different multi-receptor activity and a clinical relevance since EEG changes had been strongly related to positive clinical outcome in schizophrenic patients.
This was a small study, using only eight animals. According to the author, however, a limited number of samples can be used when applying the QEEG technique to characterise the PK / PD profiles of early compounds.
Importantly, this work highlights the value of using the QEEG approach in preclinical and early phase investigation as it provides proof that a compound passes the blood-brain barrier; it allows translational approaches by comparing animal model results with human results; and provides data in the form of EEG profiles, comparing an existing compound to investigational ones. These are useful tools that help pharmaceutical sponsors make better GO / NO-GO decisions for CNS drug candidates.
Baldeweg et al. (2006) used evoked potential, specifically impaired mismatch negativity, as a biomarker of Schizophrenia in a study of the effects of acute administration of nicotine on auditory sensory memory. The study was conducted in 20 healthy smokers who were randomised to either nicotine gum or placebo. Evoked potential responses to deviations in tones (deviants) were recorded using constantly changing standard stimuli in order to measure the effect of stimulus repetitions on the encoding of new stimuli. Results showed that there was a marked effect of stimulus repetition on the standard EP. Acute nicotine administration increased MMN amplitude significantly in the treatment group as compared to the baseline recording, whereas no changes in MMN were seen in the placebo group.
Although schizophrenics were not included in this small study, the results support validating this approach as a possible surrogate marker for the treatment of cognitive deficits in Schizophrenia and with the observed functional impairment observed in patients. MMN might therefore be possible. Schizophrenia biomarkers with benchmarking of ‘novel therapies’ assessed not only against placebo but against nicotine mediated improvement in the patients’ MMN impairment (enhancing stimulus encoding and sensory memory).
Moving beyond small, single-site studies
For the most part, the EEG, QEEG and EP methodologies are used in small preclinical and Phase I studies in the hope of gathering and comparing data in animals, healthy volunteers and CNS patients to identify promising drug candidates. As those therapies move forward, the tools have application for Phase II studies and eventually into multi-centre studies. This section discusses two clinical trials in which neurophysiological approaches were incorporated into later phase testing.
In one study, routine EEGs were performed as part of a multiple dose Phase I trial of a compound with a glutamatergic mode of action, meaning that it impacts the neurotransmitter system linked to memory formation and information processing, and to excitatory CNS effects, up to and including seizures. The therapeutic indication studied was a degenerative disorder (Alzheimer’s disease), although this neurotransmitter system is also implicated in Schizophrenia, Major Depression and other CNS disorders.
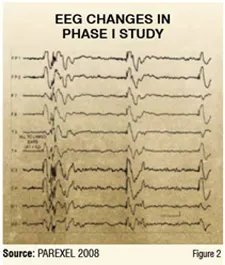
The study enrolled 12 healthy male volunteers, who were randomised to a low, mid, or high dose of the drug. The EEG changes shown in Figure 2 reflect the high dose arm two hours after drug intake in 50 per cent of the subjects. The changes show electrical discharges with high amplitudes indicating an increased likelihood of seizure. The low and midrange dosage groups did not show any changes known as epileptiform discharges, suggesting that they are safer with respect to seizure likelihood.
In a follow-up study in healthy male volunteers, a first tonic-clonic seizure appeared as a serious adverse effect. The observed EEG data led to an adjustment in dosage in consecutive Phase II studies and to the implementation of routine EEG monitoring in a central EEG reading unit.
One of the first larger studies to use QEEG as a predictive tool is known as Biomarkers for Rapid Identification of Treatment Effectiveness in Major Depression (BRITE-MD). It is being conducted in 10 locations throughout the United States (Chart 1) and sought to recruit 375 subjects with a diagnosis of major depressive disorder. The goal is to evaluate the potential of early EEGs as a predictor of an individual’s response to treatment with specific antidepressant medications. Enrollment was completed in March 2007, with 220 subjects analysed for 7-week endpoints.
The BRITE-MD trial tests two antidepressants—escitalopram (Selective Serotonin Reuptake Inhibitor (SSRI)) and bupropion XL—studying them as single-drug therapy and in combination. It also introduces a dose escalation phase. The intent of the study is prospective validation of an Antidepressant Treatment Response (ATR) indicator as a biomarker of clinical response to an SSRI, and to determine the best treatment option for patients who are predicted to be non-responders to an SSRI. The study is designed to use EEG, starting on Day 0, to determine the association between drug treatment outcome and brain function in patients with Major Depressive Disorder. Electrodes are placed on a patient’s forehead and earlobes to measure brain responses that appear within seven days, and sometimes as early as 48 hours, after beginning antidepressant treatment. On Day 7, there is an ATR assessment, clinician prediction of response, and randomisation.
Interim results indicated that the ATR index reached statistical significance in predicting seven-week clinical response as measured by the standard Hamilton Depression Rating Scale after just one week on treatment with an SSRI. Results presented in December 2007 showed ATR-predicted responders to escitalopram are significantly more likely (67 per cent) to respond to seven weeks of escitalopram treatment than are ATR-predicted non-responders (28 per cent). Findings also showed that ATR-predicted responders are significantly more likely (50 per cent vs. 21 per cent) to achieve remission with seven weeks of escitalopram treatment, and ATR predicted non-responders to escitalopram are significantly more likely to respond if switched to bupropion XL at one week (53 per cent) versus staying on escitalopram for seven weeks (28 per cent). These findings suggest that early EEG testing can detect an early brain signal of response, which can help clinicians determine treatment response much faster and effectively than a trial and error approach that can take weeks.
Going forward
Reducing the timelines and cost of investigational therapies are keys to expanding the number of treatment options for the growing number of people suffering from a range of CNS disorders. One promising approach for speeding the clinical development process is to use established neurophysiological tools, such as the routine EEG, quantitative EEG and EP, in a search for biomarkers that allow researchers to integrate preclinical results in the design of Phase I studies. How do results of first-in-human studies compare to what was seen in animal studies? Using these methods allows researchers to characterise the PK / PD profile of compounds early on and to compare results to preclinical data in an effort to determine whether a compound crosses the blood-brain barrier or produces CNS-mediated side effects in humans. Frequently, there is a platform of compounds with similar properties under development—differentiating these, one from the other, and selecting the best drug for development is critical. These electrophysiological techniques, along with imaging and cerebral spinal fluid sampling can provide invaluable information and accelerate drug development.
With EEG, QEEG and EP, small studies can be conducted in which healthy volunteers are exposed to varying doses of a compound. This approach can bring into Phase I the kinds of dose-ranging studies that traditionally occur in Phase II. Pushing early dose-ranging into Phase I and matching results to preclinical work holds promise for improving the GO / NO-GO decision-making process. In addition, these tools enable the use of surrogate biomarkers, such as whether an EEG indicates central action. Later on, if the biomarkers become validated surrogate endpoints, their use could reduce years from the overall clinical development path. By increasing detection sensitivity and reliability, samples sizes required to demonstrate efficacy would be substantially reduced. CNS drugs typically require large multi-centre global trials to achieve adequate response data against placebo, and take years to conduct. Clinical trials would benefit from the incorporation of potential bio / surrogate markers as a corroborative signal converging on the currently employed clinical endpoints, especially when the diminished placebo versus the drug effect size difference is small.
A variety of neurophysiologic techniques are being employed to study compounds early on in clinical development, in the target diagnostic CNS patient population. By bridging animal findings to humans, and then from volunteers to patients, these techniques contribute to our ability to translate preclinical disease and toxicity models to patients, and inform drug developers of possibly effective doses. Currently, these methods are being validated in small Phase I trials, but they hold promise for use in later stage studies. The infrastructure to conduct QEEG or EP in large-scale clinical trials is becoming a possibility, as equipment becomes more portable and affordable, and central reading of data is readily achieved by electronic data transmission.
References
- 10 Facts on the Global Burden of Disease, World Health Organization, http://www.who.int/features/factfiles/global_burden/en/index.html, accessed February 6, 2009.
- Ferri CP, Prince M, Brayne C, et.al. Global Prevalence of Dementia, A Delphi Consensus Study, Lancet 2005;366:2112-7.
- Labor Day 2001 Report, Untreated and Mistreated Mental Illness and Substance Abuse Costs US $113 Billion A Year, 2001, http://www1.nmha.org/pdfdocs/laborday2001.pdf, accessed February 6, 2009.
- Medicines in Development for Mental Illnesses, Pharmaceutical Researchers and Manufacturers of America 2008, http://www.phrma.org/files/Mental 2008.pdf, accessed February 6, 2009.
- Kaitin KI, editor, Longer Clinical Times are Extending Time to Market for New Drugs in US, Tufts Center for the Study of Drug Development Impact Report 2005, Nov/Dec 7.
- Pangalos MN, Schecter LE, Hurko O. Drug Development for CNS Disorders: Strategies for Balancing Risks and Reducing Attrition, Nat Rev Drug Discov 2007;6:521-32.
- Cantor C, Evolution and Post Traumatic Stress, Routledge, 2005.
- Sams M, Alho K, Na?a?ta?nen R. Sequential effects on the ERP in discriminating two stimuli. Biol Psychol 1983;17:41?58.
- Parker TJ, Della Pasqua OE, Loizillon E, et.al. Pharmacokinetic-pharmacodynamic modelling in the early development phase of anti-psychotics: a comparison of the effects of clozapine, S 16924 and S 18327 in the EEG model in rats, Br J Pharmacol 2001;132:151-8.
- Baldeweg T, Wong D, Stephan KE. Nicotinic modulation of human auditory sensory memory: Evidence from mismatch negativity potentials, Int J Psychophysiol 2006;59:49-58.
- BRITE Final Results Conference Call, Aspect Medical Systems, December 11, 2007, http://library.corporate-ir.net/library/73/737/73770/items/273305/BRITEStudyResults1207.pdf, accessed February 9, 2009






