
Adaptive Clinical Trial Designs
Esther Mahillo, Vice President, Operational Strategy and Feasibility, Precision for Medicine
Adaptive clinical trial designs are a preferred approach when certain circumstances concur, like rare diseases, a need to optimise the dose finding process in patients, or a medical open need lacking a treatment. We discuss in this interview the utility, level of implementation, and future trends in adaptive clinical trial designs.

How would you define adaptive clinical trials?
The definition, concept, and scope of controlled clinical trials in human beings is a relatively new one and has rapidly evolved during the last 100 years. Regulations and guidance on the conduct of clinical research worldwide, are based on three principles:
1) a prospective and predetermined assignment of medical intervention in human beings
2) balance between efficacy and safety of the medical intervention and
3) the voluntary and informed acceptance of subjects to participate in the investigation.
The main objective of clinical trials is to generate scientific evidence to support relevant decisions related to therapies for human beings. The strength of scientific evidence relies on two principles: the strength of the study design, and the fortress of endpoints measured. At the basis of the pyramid of scientific evidence we find the expert opinion. At the top, the paradigm of best scientific evidence is data generated on randomised, double-blind, placebo-controlled (when possible) clinical trials (RCT), aimed to demonstrate with the lower possible biases, that a new therapeutical approach is better (safer, more efficacious) than the standard available treatment. There are different guidelines on scientific evidence ranking, from the one published in United States by its Preventive Services Task Force (USPSTF) to the Oxford Center for Evidence-Based Medicine (EBM) Levels of Evidence. The Grading of Recommendations Assessment, Development, and Evaluation (GRADE) has been endorsed by over 100 organisations worldwide (gradeworkinggroup.org).
Adaptive trials differ from the classical ones in the fact that therapeutic and business decisions are taken during the study conduct, based on ongoing analysis of data. This approach is based on statistical designs which are well validated and use predefined algorithms for decisions (Krendyukov, A. et al. Value of Adaptive Trials and Surrogate Endpoints for Clinical Decision-Making in Rare Cancers. Front. Oncol., 08 March 2021).
Which are the advantages or disadvantages of adaptive clinical trials?
Some of the benefits of conducting clinical trials with adaptive designs are:
- Reduce sample sizes and total cost while maintaining statistical power
- A higher number of patients treated at optimal dose levels
- Quick and efficient identification of patient sub-populations who may better benefit from investigational treatment
- Early stop of clinical trials with ineffective and/or unsafe treatments
- Acceptance by a majority of regulatory authorities of these adaptive designs to grant conditional accelerated approval to certain drugs.
However, its counterpart is the additional level of organisational and logistical effort required, including the role of subject matter experts and the establishment of different data review committees. Most of the adaptive designs require ongoing or multiple interim analyses of data, the establishment and management of Safety Review Committees (SRC), formed by members related to the research; and/or Data Safety Monitoring Boards (DSMB), embodied by independent subject matter experts.
Additionally, more frequent and fluent communication with the regulatory authorities is needed to guarantee that the designs and study endpoints are adequate to the patients and investigational needs, the study hypothesis remains valid, and the decisions taken throughout the study are adequate.
Why is there a shift from the classical drug development pathway to adaptive clinical trials?
The clinical development plan for a new drug is generally based on traditional study designs, which first need to demonstrate that safety administration in human beings is feasible (phase I, optimal safer dose level finding); shows signals of efficacy to guarantee further investigation (phase II, proof of concept); and renders a superior result than the standard therapy (phase III, pivotal clinical trials). This classical drug clinical development plan is a lengthy process that can take as long as 10 years, and it is expensive, with a median cost of $985 million, and an average cost of $1.3 billion (Woters J. JAMA. 2020;323(9):844-853).
However, generating top-level scientific evidence is not the end of the process, but the starting point for the regulatory agencies to assess the data, and integrate all other criteria which may influence their decision-making, like quality of life, availability of similar drugs, pharmaco-economy data, etc. At this stage, a paradox can happen that a new therapy has met the golden criteria for scientific evidence level I, but the data, though statistically significant, may not be clinically meaningful. For example, a phase III, RCT comparing the investigational product to the best standard of care, proves superior efficacy with a longer median overall survival in a cancer population. The design and the primary endpoints are the best to guarantee the highest level of evidence. But the magnitude of improvement between earlier median overall survival and new one is in the range of a few days… In this context, the level of recommendation for the new drug is expected to be low, the same as the probability to get this drug approved by the regulatory authorities.
The average time for review of the informative dossier for a new therapy varies depending on the country or region involved and may range from 6 to 12 months in US or European Union, (health.ec.europa.eu; www.drugwatch.co), and has been historically longer in some Asian countries, like China.
Under certain circumstances, all sponsors, patients, and society may benefit from a more rapid and efficient approach to classical drug development.
How are adaptive designs used to shorten clinical drug development?
The classical clinical development pathway may become suboptimal in certain scenarios. For example:
- Patients are in a dire need for a therapy, in diseases where no efficacious treatment has yet become available
- Rare indications, with a small number of subjects affected globally. The epidemiology data may not support a classical study design which may require hundreds or even thousands of patients
- Patients with potentially deadly diseases, who need new therapies to change expectancy of life, etc.
Regulatory authorities have embraced the commitment to accelerate the approval process of new drugs in such cases. For example:
- FDA, through its Center for Drug Evaluation and Research’s (CDER)
- EMEA, thought the Medicine´s Adaptive Pathways to Patients (MAPP) initiative
- Australian Therapeutic Goods Administration (TGA) through its Priority Review Pathway; etc.
However, we still need scientific evidence based on study design and strength of study endpoints. The response is an “intermediate step” in the clinical development pathway, where we seek a conditional marketing authorisation to allow early access of patients to more efficacious drugs, while the mature data is obtained.
Adaptive designs in clinical trials are part of these solutions, and aimed to shorten drug clinical development, while minimising the number of subjects exposed during the research.
Are adaptive clinical trial designs beneficial in proof-of-concept (phase II) studies?
Phase II studies may benefit from an adaptive design under certain circumstances such as in biomarker-driven pathologies, where the presence of a biomarker implies a worse prognosis and/or can be predictive of response to targeted therapies. Certain study designs are aimed to select only those patients with a biomarker-positive disease, to test the potential efficacy of targeted therapies against this biomarker. A similar principle applies also in the research of gene therapies with genetic mutations.
Some of the more frequent study designs used in this context are:
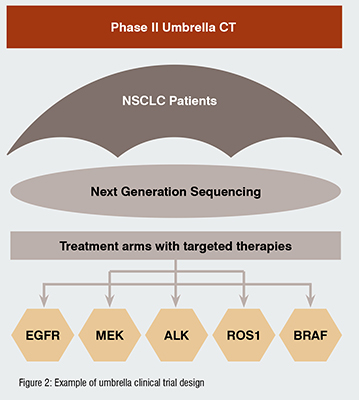
UMBRELLA STUDIES: evaluation of multiple investigational products in patients with same primary disease, characterised by expression of different biomarkers. For example, patients diagnosed with non-small cell lung cancer (NSCLC), can present different altered genes, like EGFR, ALK, ROS1, BRAF, NTRK, MET, or RET. A phase II umbrella study in NSCLC would be based on genotyping of patients, and assignment to biomarker-driven targeted therapies: (Figure 2)
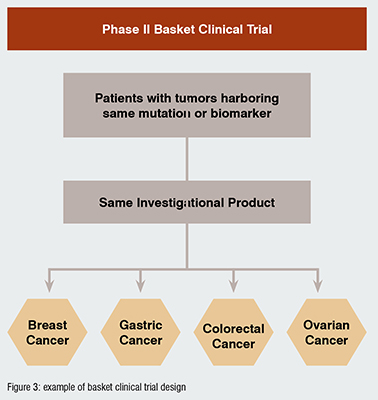
BASKET STUDIES: evaluation of one targeted therapy in patients with different indications, which have in common the expression of the same biomarker. For example, overexpression of HER2 has been observed in different tumour types, like breast, gastric, and colorectal cancers, etc. A basket study would test one single targeted anti-HER2 drug in multiple arms enrolling different patient populations to test individual efficacy on each one of them and make decisions on study design for phase III RCT. (Figure 3)
Among the different adaptive approaches, bridging clinical trials (phase I/II, phase II/III), allow for ongoing re-definition of the target patient population and decision making (stop or continue).
Are adaptive clinical trial designs used in early phase studies?
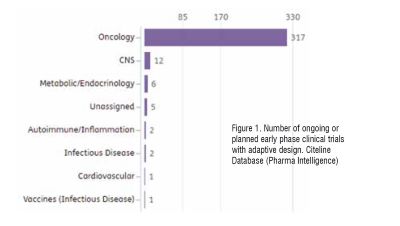
Adapted study designs in early phase studies are broadly accepted and implemented, with oncology leading the ranking of planned or ongoing clinical trials, as per Citeline data (search date: April 2023) (Figure 1)
In oncology, the classical 3+3 dose escalation design was originally used with cytotoxic drugs, and the principle to make decisions on escalating or de-escalating the dose is based on maximum toxicity in 33 per cent of patients deemed as acceptable. This model has evolved with alternative algorithm-based designs such as accelerated titration, Bayesian optimal interval (BOIN), continual reassessment method, escalation with overdose control, or modified toxicity probability interval (mTPI-2), among others (doi: 10.3390/cancers14061566).
A combined phase I (dose escalation)-phase Ib (selection of target subpopulations)-phase II (efficacy data obtained) is a relatively frequent approach in certain therapeutic areas. Approximately 1 per cent to 2 per cent of all ongoing or planned clinical trials in phase I, phase I/II, or phase II are using this approach (Citeline search date: April 2023).
FDA has released a guidance on this topic which may be used as a reference: “Expansion Cohorts: Use in First-In-Human Clinical Trials to Expedite Development of Oncology Drugs and Biologics (August 2018)”.
Are adaptive clinical trial designs used in pivotal (phase III) studies?
In later phases of development, an adaptive design may serve different objectives, like sample-size re-estimation, dynamic modification of the primary endpoint, closing or maintaining study arms, or setup of multiple investigational combinations, during the conduct of the study. There are varied examples of these approaches, and some of them are considered as standard in some areas.
What is the present (and future) of adaptive clinical trials?
Adaptive clinical trial designs in an early phase of development (phase I, phase I/II, phase II) are consolidated in some therapeutic areas, where sponsors and patients need rapid decisions based on critical safety and efficacy data, which can be obtained with a lower number of subjects exposed at a more optimal dose level.
Oncology and rare diseases have been the areas where adaptive trials have been more frequently used to support accelerated conditional approval of new drugs. A certain number of them have been obtained with results of single-arm studies, utilising intra-arm or historical data as controls. Whereas this approach seems to be still valid and well received by the regulatory authorities for some rare diseases, the FDA released in March 2023 a new guidance for oncology research (Clinical Trial Considerations to Support Accelerated Approval of Oncology Therapeutics Guidance for Industry), which supports the RCT design as the golden rule to obtain scientific level 1 data, and therefore, facilitate accelerated approval granted.
In conclusion, the arena of adapted clinical designs is a dynamic environment and close attention should be paid to this evolving field to obtain the maximum benefits of them.
Author Bio

Mahillo has 30 years of experience in clinical research. She is a post-grade magister at 4 Spanish Universities and the author of 3 books, 25 scientific articles, and 27 contributions to congresses. She obtained the first prize from the SEOM for her work on enhancing clinical research awareness among patients











