
The pharmaceutical industry continues to face challenges in improving its R&D cost-effectiveness. Despite the steady increase in the R&D expenditure in the U.S. pharma industry over the past 10 years, from US$ 17.5 billion to US$ 55.3 billion, the number of new products launched has decreased from 53 in 1996 to 22 in 2006 (Price Waterhouse Coopers, Pharma 2020, 2006).
There is not a single solution to the problem, but improving the quality of the exploratory development candidates selected to go into confirmatory clinical development should further reduce the risk of late-stage failures.
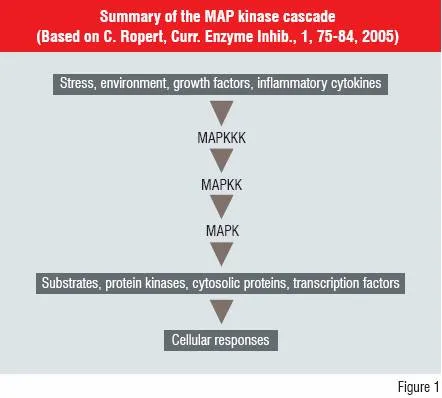
Target complexity and selection of outcome measures
Many new drug discovery targets have been influenced by information from sequencing of the human genome. However, they may complicate the understanding of the physiological outcomes of modulating biological activity via these new targets. An example of the complexity is shown for the Mitogen Activated Protein (MAP) kinase cascade (Figure 1).
The MAP kinases are involved in coordinating the activation of gene transcription, protein synthesis, cell-cycle activities, cell death and differentiation. The three layers of kinase activity modify the expression of a series of downstream products. This cascade can affect the innate immune response and offers potential routes to modulate inflammatory diseases such as Rheumatoid Arthritis.
However, modulating such pathways may mean the clinical side-effect profile and safety of drugs acting on such new targets cannot be predicted accurately by non-clinical studies. An improved understanding of the relationships between the target, physiological effects, pharmacology and toxicology is needed, increasing the focus on the search for and evaluation of biomarkers.
Biomarkers, surrogate and clinical endpoints
These parameters were defined by the Biomarkers Definitions Working Group in 2001 as follows:
- A biomarker is a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes and/or pharmacologic responses to a therapeutic intervention
- A surrogate endpoint is a biomarker that is intended to substitute for a clinical endpoint e.g. HbA1c
- A clinical endpoint is a characteristic or variable that reflects how a patient feels, functions or survives
In some therapeutic indications there are no easily measurable clinical endpoints available and biomarkers may be used to support Go/No go decision. Therefore, novel biomarkers are needed that can:
- serve as indicators of potential toxicity or efficacy to support early Go/No go decisions either in lead candidate selection or in early clinical studies
- support patient inclusion/exclusion in clinical trials and support the submission for regulatory approval
- provide a diagnostic to identify the patients most likely to benefit from treatment with the drug (individualised medicine)
Various biomarkers have been used in the evaluation of drug activity and safety, and no single biomarker can provide all the desired information. Some have been available and used for a long time e.g. clinical blood biochemistry and haematology parameters such as liver enzymes creatinine and Hb. However, the application of new biomarkers must inevitably be empirical, as selection, measurement and evaluation of their relevance proceeds parallely.
The effects of drugs on in vitro, ex-vivo and in vivo lipopolysaccharide stimulated release of IL-1?, TNF?, IL-6 have been used to assess potential anti-inflammatory activity, and provide early mechanistic proof of concept for some kinase inhibitors. In some cases, such techniques can also provide an indication of the potential to disrupt normal immune responses.
Drug induced changes in gene-expression in vitro or in vivo can provide early indications of mechanistic proof of concept and/or toxicity. Huang et al. investigated the expression of 684 genes in rat liver following acute oral hepatotoxic doses of acetaminophen, methapyrilene, furan, methotrexate and phenytoin. Principle Component Analysis (PCA) of the expression patterns was able to distinguish the different hepatotoxic changes caused by these compounds and also estimate the type and severity of the toxicity.
As with toxicogenomics, investigation of the expression patterns of tissue proteins can indicate the potential toxicity of new drugs. Petricoin et al. successfully used proteomic analysis of low molecular weight proteins (<15>
Depletion of glutathione in vitro or in vivo is an indicator of the production of reactive species e.g. drug metabolites, products of auto-oxidation and may be used as an early indicator of an increased risk of toxicity in early non-clinical evaluation. Such biomarkers can help identify wanted and unwanted effects early in drug candidate selection.
Biomarkers - answers or questions?
There is no simple answer as many of the disease-modifying targets involve multiple processes and interactions in the target species. The value of the information provided will depend on the understanding of the relationship between the biomarker(s) and the wanted or unwanted effects. This is more difficult with newer biomarkers where such relationships are being evaluated in parallel with their non-clinical and clinical applications.
Thus, the p38 MAP kinase inhibitors’ concentration-effect relationships can be studied in vitro in human blood using the addition of LPS to stimulate the release of a pro-inflammatory cytokine e.g. IL-1?, IL-6 and/or TNF? in samples containing different concentrations of the inhibitor. The experimental conditions e.g. whole blood, diluted whole blood, measuring effects on specific cells and the cytokine stimulant concentration e.g. LPS in vitro, ex-vivo or in vivo can influence the intensity of response in controlled and treated samples and hence the results and their interpretation.
Depending on the biomarker being measured and the target indication, the relationship between the drug concentration-response curves may well be different in blood from healthy volunteers and patients. This may influence the selection of the population for the initial clinical investigation i.e., healthy volunteers or patients. Therefore, careful consideration of the experimental conditions used in vitro, ex-vivo and in vivo and target population is important in biomarker application.
Microdose studies in Go/No go decision-making
Microdose studies in man are being used more frequently to support Go/No go decisions. This approach uses a dose not expected to produce any pharmacological response, with a maximum dose of 100?g, to define aspects of the clinical Pharmacokinetics (PK) of novel drug candidates early in development. These studies require more limited non-clinical safety evaluation and a number of analytical techniques have been used:
- • Highly specific, high-sensitivity (LLOQ ~ 10-15 g/ml) LC-MS/MS or GC-MS methods for drug and/or metabolite(s)
- • Accelerator mass spectrometry (AMS; LLOQ ~ 10-18 to 10-21g/ml) with a carbon-14 labelled drug microdose (? 100?g; nCi)
- • Positron Emission Tomography (PET; LLOQ ~ 10-15g/ml) with a carbon-11 labelled drug microdose (? 100?g; nCi)
AMS and PET detect and quantify drug-related material in biofluids (plasma, urine) and/or tissues measuring total radioactivity, unless combined with chromatography. Combining them with a specific LC-MS/MS method for the drug can increase the value of the PK data. The method(s) used should be influenced by the question(s) to be answered, in order to obtain the required information as efficiently and cost-effectively as possible.
PET studies have been used to investigate blood-brain barrier penetration to support the selection of Central Nervous System (CNS) drugs and AMS has been used to select drug candidates with appropriate PK characteristics e.g. absorption, clearance and half-life in microdose studies.
The extent to which microdose PK predicts those at pharmacological doses was investigated by Lappin et al. in an AMS study. The PK obtained in a microdose study were in good agreement with those at pharmacological doses for diazepam, midazolam and ZK253. The clearance, but not volume of distribution of warfarin, showed reasonable agreement. Microdose data could not be obtained for erythromycin, possibly because of degradation in stomach acid.
Microdose studies are finding increasing application in support of early candidate selection. However, they will not always be successful in predicting PK, and do not allow any PK/PD investigations.
Biofluid metabonomic analysis
Biofluid analysis has been used extensively to evaluate non-clinical and clinical safety. Exposure of a biological system (cells in culture, animals or man) to a toxin changes the expression of various genes, biosynthesis of various proteins and ultimately results in changes to endogenous metabolic pathways.
The effects are dependent on the nature of the toxic challenge, but occur at different times after the toxic insult, with the metabolic changes tending to appear later than gene expression and protein biosynthesis changes.
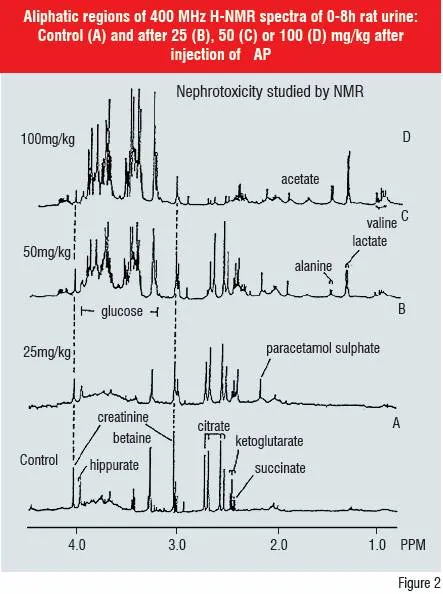
Direct biofluid H-NMR, LC-MS and LC-MS/MS have been used in metabonomic screening. H-NMR has been shown to be a valuable indicator of toxicity based on pattern recognition of time-dependent changes in endogenous metabolite concentrations e.g. creatinine, lactate, glucose, ?-ketoglutarate in plasma and/or urine. This metabonomic approach has been defined as “the study of multi-variate, time-resolved metabolic changes in biofluids to patho-physiological insult or genetic modifications in cells, organs and the whole animal.”
The biofluid Nuclear Magnetic Resonance spectroscopy (NMR) technique has shown that specific changes occur in endogenous metabolite profiles dependent on the organ affected e.g. kidney, liver, testis and the site of toxicity in the target organ e.g. proximal or distal renal tubule toxicity in the kidney. Single and repeated dose toxicity and its reversibility can be studied with the added benefit that the nature of the metabolic change does not have to be known in advance.
A collaborative project using some 80 known toxic compounds has confirmed and extended previous experience on identifying biomarkers of liver and kidney toxicity in rats and mice. Biofluid H-NMR clearly identified control, low and high dose animals and, between-laboratory differences were much smaller than those between animals and doses.
Experience with H-NMR biofluid analysis suggests that it should be used more extensively and consistently in support of early non-clinical and clinical evaluation.
Reducing risks of hepatotoxicity in man
Liver toxicity is seen as the most serious drug-induced adverse event in clinical application of drugs and in the USA some 40 percent (patients >50 years) and 10 percent (all age groups) of liver disease is attributed to drugs.
Hepatotoxicity has stopped the development or marketing of several drugs including troglitazone, bromfenac, tienilic acid and temofloxacin and resulted in restricted use or withdrawal in some countries of zileuton, trovafloxacin, tolcapone and felbamate.
Non-clinical safety testing detects 50 percent of observed human liver toxicities. Although there is a good correlation across species allowing detection of intrinsic hepatotoxins, detection of idiosyncratic hepatotoxins has proved more difficult. Such toxicities occur in less than five percent of treated subjects, appear unrelated to the pharmacology of the drug, lack a clear dose response and show inconsistent patterns of appearance with time.
They are difficult to detect in non-clinical or in Phase III clinical studies, because a low incidence (e.g. 1/1000) would require a study with some 18,000 subjects. The possible mechanisms that could lead to idiosyncratic hepatotoxicity and potential indicators of the risk of such effects have been discussed.
Some indicators of potential idiosyncratic hepatotoxicity
Several characteristics appear to be associated with the potential for idiosyncratic reactions, including structural similarities to other hepatotoxins, glutathione depletion, glutathione conjugate formation and covalent binding of drug/metabolites to cellular macromolecules e.g. at levels ?50pM. In addition, hepatic bioaccumulation, potential for drug-drug interaction, evidence of liver toxicity, liver hypertrophy and/or P-450 induction with no safety margin in non-clinical toxicity, were also considered as indicators of this potential. Drugs exhibiting several of these characteristics seem to be at more risk of producing idiosyncratic hepatotoxicity.
Improving in vitro non-clinical test systems - Hepatotoxicity
Drug metabolism e.g. CYP-450 oxidation, glutathione or glucuronic acid conjugation may result in hepatotoxicity via reactive metabolites. These may cause immune-mediated toxicity by covalent binding of a reactive metabolite with an endogenous protein e.g. practolol. Oxidative stress, glutathione depletion, damage to cellular proteins/membranes, and/or lipid peroxidation are also routes to hepatotoxicity.
Whether such effects result in hepatotoxicity may depend on the extent to which they occur in association with a possibly unrelated predisposing inflammatory reaction and this added complexity may contribute to the low incidence of such idiosyncratic responses.
Various in vitro test systems including primary hepatocytes, immortalised cell lines, liver slices and non-clinical toxicology studies are used to assess hepatotoxic risk.
Dambach et al. used four separate primary human hepatocyte-derived immortalised cell lines. These cell lines had low intrinsic P-450 and glucuronidation capacity, but were genetically modified to express some 70, 30, 95 and 20-fold higher levels of CYP-450 3A4, 2C9, 2C19 and 2D6 respectively. The intention was to enhance the ability of such a system to produce and detect toxic i.e. reactive metabolites, while having qualitatively similar metabolite profiles to those of normal primary human hepatocytes.
These cell lines were used to measure concentration-dependent cytotoxicity in a multi-tiered screen of acute direct or metabolism-dependent hepatotoxicity.
This system was validated by comparing in vitro predictions with the observed hepatotoxicity for 679 marketed drugs. The system correctly predicted 585 of 587 non-hepatotoxic drugs, 15 of 21 severely hepatotoxic and 51 of 71 variably hepatotoxic drugs. The system scored false negatives for six severe, non-CYP450 dependent, hepatotoxins, and failed to detect 20 hepatotoxic drugs acting via cholestatic mechanisms.
This system was subsequently used to screen novel compounds. Those with a cytotoxic IC50 ? 50?M in any cell line were considered to have an increased hepatotoxic risk and were either stopped or subjected to further investigation. The relationship between IC50 and over-expressed CYP-450 indicated the importance of metabolism in the toxic pathway.
Selected drugs tested in this system also showed changed expression of 40 genes and two proteins that distinguished an idiosyncratic hepatotoxic drug (troglitazone) from negative control and six non-hepatotoxic drugs.
Alden et al. used some 20 genes associated with inducible metabolism and inducible toxicity to screen for hepatotoxicity in vitro. This system correctly identified many classical hepatotoxic drugs, two known idiosyncratic hepatotoxins (zileuton and troglitazone) and negative and positive control drugs when tested at 1 and 5?M.
Such hepatotoxic screens should be used as part of an integrated approach taking account of structure-activity data, cytotoxicity and/or enzyme induction in primary hepatocytes, metabolic pathways, and in vivo safety data. There is no simple way to select better development candidates. However, these are some of the techniques available to screen compounds for wanted and unwanted effects, as part of an integrated strategy to improve decision-making and help improve the
success of pharma R&D.
References
• Price Waterhouse Coopers, Pharma 2020, 2006
• C. Ropert, Curr. Enzyme Inhib., 1, 75-84, 2005.
• L. Chang and M. Karin, Nature, 410, 37-40, 2001.
• Biomarkers definitions working group, Clin.Pharmacol. Ther., 69, 89-95,2001.
• F. Goodsaid, Preclinica, 2, 259-261, 2004.
• D. A. Parasrampuria, P. de Boer et al, J. Clin. Pharmacol., 43, 406-413, 2003.
• J. Branger, B. van den Blink et al, J. Immunol., 168, 4070-4077, 2002.
• Q. Huang, X. Jin et al, Mutat. Res., 549, 147-167, 2004.
• E. F. Petricoin, V. Rajapaske et al, Toxicol. Path., 32 (Suppl. 1), 122-130, 2004.
• C. W. Vose, Drug Metab. Pharmacokin., 20, (Suppl 10, 23-27, 2005.
• M. Bergstrom, A. Grahnen and B. Langstrom, Eur. J. Clin. Pharmacol., 58, 357-366, 2003.
• R. C. Garner, Curr. Drug Metab., 1, 205-213, 2000.
• G. Lappin and R. C. Garner, Anal. Bioanal. Chem., 378, 356-364, 2004.
• G. Lappin, W. Kuhnz, R. Jochemsen et al, Clin. Pharmacol. Ther., 80, 203-215, 2006.
• J.K. Nicholson, J. Connolly et al, Nature Reviews Drug Discovery, 1, 153-161, 2002.
• J. K. Nicholson, J. C. Lindon and E. Holmes, Xenobiotica, 29, 1181-1189, 1999.
• J. Gartland et al, J. Pharm. Biomed. Anal., 8, 963-968, 1990.
• J. Gartland et al, Mol. Pharmacol., 39, 629-642, 1991.
• J. Lindon, J.K. Nicholson et al Toxicol. App. Pharmacol, 187, 137-146, 2003.
• C. Alden, J. Lin and P. Smith, Preclinica, May/June, 27-35, 2003.
• T. Peters, Toxicol. Path., 33, 146-154, 2005.
• P. Greaves, A. Williams and M. Eve, Nature Reviews, 3, 226-236, 2004.
• K-U. Petersen, Arzneim. Forsch., 423-429, 2002.
• D. M. Dambach, B. A. Andrews and F. Moulin, Toxicol. Pathol., 3, 17-26, 2005.





