Since the introduction of the ICH E6 (R2) addendum, the industry has seen a rise in the implementation of a variety of risk-based monitoring (RBM) methodologies and technologies. However, quite often, small and mediumsized enterprises (SMEs) are more reluctant to adopt such methods as they perceive these types of approaches to be expensive and complicated. Consequently, they are generally unsure of the steps they need to take to implement an RBM strategy. As the industry changes and evolves, it will be essential for SMEs to learn how the regulations will impact them, and to understand how an RBM approach can be implemented quickly and efficiently to suit their specific needs. There are solutions out there that facilitate ICH E6 (R2) compliance and can support SMEs from pre-study risk assessment through to database lock and submission. The key is to find an approach that offers both consultation services and technological software that addresses the challenges faced by small and mid-sized research organisations in effectively adopting RBM and ensuring compliance with ICH E6 (R2).
The complexity and size of clinical trials has intensified over recent years alongside mounting costs and escalating regulatory pressures. The introduction of the finalised ICH E6 (R2) means that organisations across the industry are now reviewing the update to grasp its implications, including evaluating the requirement for modified strategies and operating models. Watching and waiting is no longer an option when it comes to RBM. Those that had not previously prioritised an RBM implementation are now driven to more determinedly assess this concept and determine an effective roll-out strategy.
Many SMEs are unsure of how to embrace RBM and often how it might add significant value. Rather than fearing the change, organisations should see this as an opportunity to embrace improved and more efficient approaches to trial design, conduct, oversight, recording, and reporting, while continuing to ensure human subject protection and the reliability of trial results. The most logical place to start is to find an expert who can provide consultation on the ICH E6
(R2) addendum and RBM methodology. The aim of a good consultation is to help organisations fundamentally understand what the regulation wants. By demonstrating how a risk-based approach meets the compliance obligations, many SMEs will be reassured that they can implement an approach that not only achieves regulatory compliance but also improves both cost and resource efficiencies.
The ICH E6 (R2) guidance specifies that the execution of RBM, and more specifically using centralised statistical monitoring (CSM) as a core component of clinical trial execution, is recommended due to its ability to provide “additional monitoring capabilities that can complement and reduce the extent and/or frequency of on-site monitoring and help distinguish between reliable data and potentially unreliable data.”1 Ultimately, the responsibility for the quality and integrity of the trial data always resides with the sponsor although a sponsor may transfer any or all of the sponsor's trial-related duties and functions to a CRO. Both the regulators and Good Clinical Practice (GCP) are making RBM an essential, and GCP compliance concern as well.
RBM and Quality by Design (QBD) have been intensely recommended not only in the updated ICH GCP guidance but in related guidance documents issued by the FDA and EMA over the past several years. QBD and RBM working hand-in-hand have the potential to yield higher quality, reduce time lines and drive greater operational efficiency in clinical research. QBD and RBM both necessitate ongoing assessment and mitigation of operational risk. QBD is conducted at the earliest stages of clinical research design to ensure that studies are optimised not just for scientific merit, but for operational success as well. Once a study protocol has been developed, QBD becomes RBM. Risk assessment is performed on completed designs by a cross-functional study team. Remaining operational risks are identified and prioritised, and risk mitigation and risk monitoring recommendations are established to guide all downstream operational study management plans.
Essentially an RBM approach includes the following:
• Critical process and data identification: processes and data that are critical to ensure human subject protection and the reliability of trial results should be identified during protocol development
• Risk identification: sponsors should identify risks - at both system and clinical trial level – critical to trial processes and data up front
• Risk evaluation: researchers must carry out risk assessment/critical data identification before the study to characterise risks through risk evaluation, looking at the likelihood, detectability and impact of those risks. They should then put together a quality oversight plan that is a targeted approach to operational quality management, specifically focused on the biggest risks to operations
• Risk control: the sponsor should decide up front which risks to reduce and/or to accept. Predefined ranges within which various quality measures will be accepted should also be established, to identify issues that can impact subject safety or the reliability of trial results. Detection of deviations from these predefined limits should trigger an evaluation to determine if action is needed
• Risk communication: quality management activities should be documented and communicated, to facilitate risk review and continual improvement during study execution
• Risk review: risk control measures should be reviewed intermittently to check that the implemented quality management activities remain effective and relevant
• Risk reporting: reporting should include a description of the quality management approach taken and review key deviations from the predefined quality limits, and any remedial actions taken.
A successful RBM centralised monitoring implementation can provide three essential aspects of value. It will make a substantial positive impact on quality outcomes across a business, resulting in additional successful marketing submissions and faster time-to-market. It also offers a practical opportunity to appraise on-site monitoring that can lead to considerable and direct savings in the cost of clinical research and clinical trial budgets. Effective centralised monitoring can also result in shorter study time frames.
The implementation of a successful centralised monitoring plan will help SMEs manage the shift towards RBM, while achieving compliance with the new regulatory guidelines. By supporting organisations in bringing significant improvements to the quality of their data, and therefore the success rates of their trials, it will be a vital tool in optimising operational quality monitoring within this exciting new paradigm for clinical research.
CSM uses statistical methods to identify unexpected or unusual patterns in clinical data. It not only drives significantly better-quality outcomes but offers greater operational resource efficiency, enabling a significant reduction in the reliance on source data verification (SDV) and related on-monitoring reviews. CSM is ideally composed of at least the following components:
• Statistical Data Monitoring (SDM): A well-designed, robust set of statistical tests to be run on all clinical data in the study, with the purpose of identifying atypical data patterns that may represent operational risks of various types including fraud, study equipment malfunction, site sloppiness and training issues.
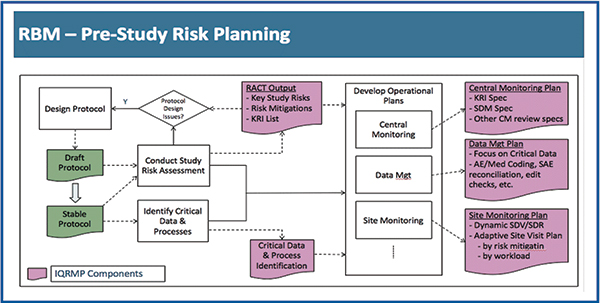
• Key Risk Indicators (KRIs): KRIs represent a set of metrics designed to help monitor for known operational risks across all sites in a study. A few examples of commonly-used KRIs include:
i. The rate of protocol deviations
ii. The rate of adverse event reporting
iii. Timeliness of data entry
iv. Rates of queries or data errors
v. Screen failure rate and early termination rate
vi. Rate of missed procedures – especially key efficacy or safety procedures
• Quality Tolerance Limits (QTLs): Similar to KRIs, QTLs represent metrics designed to monitor for specific operational risks. However, the focus is on more systematic issues, monitoring for specific thresholds beyond which the study would likely be considered an operational failure.
As a starting point, SMEs should review the below pointers to fast-track their ICH E6 (R2) compliance and RBM adoption:
• Develop an understanding of the ICH E6 (R2) guidelines and the fundamentals of RBM methodology
• Establish an overview of the pre-study risk assessment and mitigation planning process
• Establish Key Risk Indicators (KRIs) – review their purpose, key considerations for optimal effectiveness and review a set of standard KRIs
• Develop a Data Quality Assessment (DQA) – this includes comprehensive statistical data monitoring as a next step in risk detection — evaluate what it comprises and its critical importance in surfacing operational risks that may not have been anticipated via pre-study risk planning.
When selecting a Risk-Based Monitoring solution, key software elements should be incorporated, providing the necessary tools for organisations to start implementing a risk-based approach without over complicating the process, giving users the freedom and opportunity to implement further RBM activities as and when required. This can include tools that facilitate the development of risk assessments, key risk indicators, patient profiles, tracking systems and training.
While compliance is a justifiable motivator, a risk-based approach to clinical trial management should deliver much more value than simple compliance, including:
• Substantial reduction in the cost of clinical development, primarily due to the reduced reliance on 100 per cent SDV and frequent on-site monitoring visits. By way of example, in a typical Phase III trial, over 30 per cent of the average US$100 million trial cost is spent on on-site monitoring. 50 per cent of this time is spent conducting Source Data Verification (SDV), amounting to an average cost of US$15 million per trial. Even reducing this by 20 per cent using CSM results in a saving of US$3 million.
• Shorter study timelines –propelled by enhanced enrolment and retention rates, as well as more effective database lock processes
• Higher marketing approval rates, driven by significantly higher study and data quality.
Now is the time to investigate how a shift in mindset, from simply ensuring compliance towel coming valuable RBM, can offer these immense business opportunities. The ICH E6 (R2) addendum has the capacity to profoundly revise how clinical research is managed. Risk-based trial design and quality management will, undeniably, be a necessary element of the clinical research landscape for years to come.
References
1.https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R2__Addendum_Step2.pdf

Patrick holds a Marketing degree from the University of Newcastleupon-Tyne, UK, and a post-graduate Marketing diploma in Business-to-Business Marketing Strategy from Northwestern University - Kellogg School of Management, Chicago, Illinois. Responsible for leading global sales, product, marketing, operational and technical teams throughout his career, Patrick is a Senior Executive with over eighteen years international commercial experience within life sciences, healthcare and telecommunications

Prasanthi Sadhu, Editor, Pharma Focus Asia













