
The pharmaceutical industry is undergoing a transformation. It is transitioning from a closed, internal, integrated discovery and development business model generating blockbuster drugs by large pharmaceutical companies to a more open, external, collaborative model where drug products are more diversified, treatments are personalised, demand variability is higher and cost pressures are greater (Sadat et al., 2014).The biopharmaceutical industry is thriving. Sales are projected to reach US$450 billion by 2019 (Hong et al., 2017) with mAbs comprising the largest selling cohort. Biopharmaceuticals accounted for 5 per cent of total biopharmaceutical and pharmaceutical revenue in 2011 (BPOG RTM-Overview Document, 2017) but are expected to account for more than 50 per cent by 2020 and will comprise ~80 per cent of the drug development pipeline. The small molecule pharmaceutical industry is experiencing challenges and stresses due to the expiry of blockbuster patents and the dearth of new drug discoveries. Large pharmaceutical companies are addressing this development by primarily focussing on biopharmaceutical discovery and development activities and outsourcing small molecule development and manufacture to CDMOs.
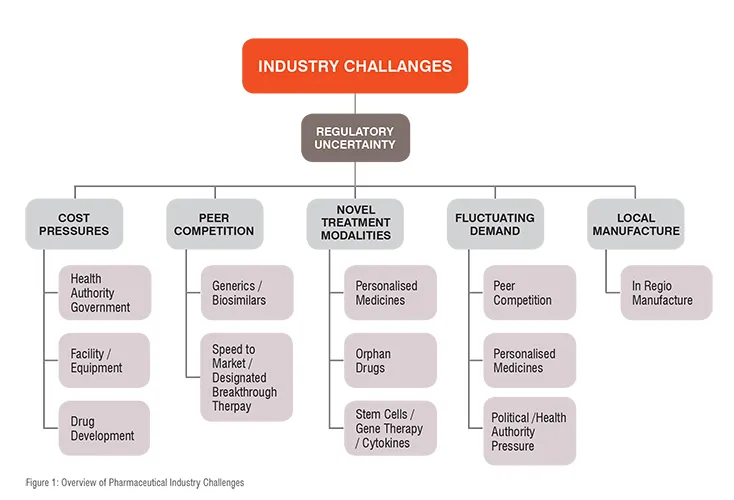
Challenges facing the industry include drug patent expiration, increasing competition from generics, biosimilars and biobetters, increasing pressure from governments and health agencies to lower the cost of drug products, the requirement for in-regio manufacture of drug products and variable demand for products. Drug discovery and development is becoming evermore demanding and costly with payers (governments, health insurers etc.) requiring companies to demonstrate that their drug is more effective than currently available drug products. The US 21st Century Cures Act (2016) mandated the FDA to assess real world data (RWD) when evaluating drug products. This has resulted in need for larger, more comprehensive and expensive clinical trials for the launch of new drugs. A summary of the challenges currently facing the industry is shown in Fig. 1.
The industry is cognisant of the pressures and is responding. Merger and acquisition activities have increased, for example, this year saw Bristol Myers Squibb’s US$74 billion acquisition of Celgene and Abbvie’s US$63 billion acquisition of Allergan. Other strategies include investing significantly in research and development, continuous improvement programmes for drug discovery, development and manufacture and greater engagement with payers and patients. Key attributes for manufacturers are the requirements to be lean, efficient and flexible and the primary business drivers identified are flexibility, speed, quality and cost (BPOG,TRM-Overview Document, 2017). Technology advances are playing a significant role in transforming the industry. Process Analytical Technology (PAT) applications are well established in the small molecule manufacturing industry primarily in the drug product area where spectroscopic technologies (IR, NIR) enable increased product throughput and improved quality. In the biopharmaceutical industry single use technologies are facilitating increased manufacturing flexibility and improved efficiencies. Levels of automation within the industry are increasing, predominantly in upstream manufacturing where in-line and on-line sensors monitor and in some applications, regulate manufacturing progress. Typical critical process parameters monitored include temperature, dissolved oxygen, pH, agitator speed etc. Readings from the sensors are used in feedback-feed forward algorithms which control the manufacturing process. Sensor accuracy and robustness has significantly improved over the past ten years resulting in more widespread adoption. Downstream manufacturing has fewer in-line and on-line monitoring applications as a result of the more dynamic environment and fewer numbers of process critical quality attributes. More recent developments involve the application of multi-parameter sensors such as Raman which can monitor multiple parameters simultaneously. Readings from these sensors are information rich and feed into multi-variate models which are employed to provide enhanced predictive process control.
Sensor technology capabilities and performance continue to improve, which aligned with advances in automation software, is supporting the introduction of continuous manufacturing within the industry. FDA considers Continuous Manufacturing (CM) to be ‘a process in which the input material(s) are continuously fed into and transformed within the process, and the processed output materials are continuously removed from the system’, where the system is defined as two or more integrated manufacturing units (FDA 2019). There are a number of modes of CM:
- Fully continuous mode: each processing unit operation is connected and run continuously. Also known as Fully Integrated Continuous Mode
- End to end continuous mode: inputs and outputs of the process are run continuously but the intermediate processing unit operations can be run in batch mode
- Hybrid mode: some parts of the process are operated in a continuous mode whereas others are run in a batch mode
- Semi-continuous mode: the manufacturing process is run continuously for a defined short period of time
- Truly continuous mode: The manufacturing process is operated continuously full time with no product hold up times..
CM is agnostic of scale and can be effectively employed in processes manufacturing drug product from kilograms to tonnes. CM generates many advantages over batch manufacturing. These include more consistent and higher quality product as in-process monitoring can detect potential process deviations in real time and apply corrective measures to correct the deviation. CM also facilitates lower manufacturing costs for example capital expenditure (CAPEX) is significantly lowered when employing multiple 2,000 litre Single Use Bioreactors (SUB) in lieu of 20,000 litre stainless steel reactors and operational expenditure (OPEX) is reduced with highly automated manufacturing trains. Other advantages include greater manufacturing flexibility, as the lower manufacturing footprint associated with SUBs and CM enables quicker construction than traditional batch manufacturing trains and also facilitates set up in a greater number of locations and markets due to the lower CAPEX and utilities requirements. The potential elimination of scale-up challenges when transitioning from clinical to commercial manufacture whereby increasing capacity is effected via scaling-out i.e. adding additional, parallel, small scale reactor trains rather than increasing reactor volume, is another significant benefit of this mode of manufacture.
CM has the potential to be a transformative technology for the industry. However, there are a number of challenges to be addressed and overcome for this potential to be realised.
Regulatory Uncertainty
Regulatory authorities have long recognised the advantages of and consistently advocate for CM. As far back as 2002 when FDA announced an initiative titled ‘Pharmaceutical Current Good Manufacturing Practices (CGMPs) for the 21st Century’, which encouraged the pharmaceutical industry to adopt new technologies and modes of operation with the objective of enhancing and improving the quality and availability of human and veterinary drugs, through the publication of ICH Q8, Q9 Q10 (2005-2008) guidance documents which introduced Quality by Design (QBD) mode of drug development and manufacture, quality risk management concepts and quality system management, to the ongoing development of ICH Q13 and Q14 guidelines (2021) which address regulatory expectations for CM and enhanced analytical procedure development which could be used to support Real Time Release Testing (RTRT) activities. In addition the ICH Q2 guideline, which addresses validation of analytical procedures, is being updated to include advanced analytical technologies which incorporate multivariate statistical analyses e.g. spectroscopic modelling. Whilst recognising the advantages of CM, the regulatory authorities acknowledge that more regulatory support and guidance is necessary to increase uptake within the industry as detailed in the ICH Q13 final business plan (2018) ; ‘The current ICH Guidelines do not sufficiently address technical and regulatory requirements that are unique to Continuous Manufacturing (CM). A harmonised regulatory guideline can facilitate implementation, regulatory approval, and lifecycle management, particularly for products intended for commercialisation internationally’. FDA in their 2019 draft guidance ‘Quality Considerations for Continuous Manufacturing’, which will form the backbone for ICH Q13, acknowledge the nascent status of CM and in the guidance state ;‘FDA recognises that continuous manufacturing is an emerging technology that can enable pharmaceutical modernization and deliver potential benefits to both industry and patients’.
In their press release announcing the draft guidance, Scott Gottlieb, the former FDA Commissioner and Janet Woodcock, Director of CDER, state CM is ‘still new and developing. Harnessing the full potential of these innovations will require us to invest time and resources in developing scientific standards and policy and supporting implementation’. This commitment to providing resources in supporting the greater uptake of CM within the industry will provide a significant boost to the implementation of CM applications within the industry. It will also require greater collaboration between the manufacturers and the regulatory agencies including sharing more information/experiences at earlier stages of implementation.
Technology Harmonisation
CM technologies such as operating platforms (MES, DCS, SCADA), sensors (temperature, DO, nutrient and metabolite monitoring etc.), sampling devices, perfusion, culture media and multivariate modelling applications have advanced considerably over the past ten years. Examples include the development of bespoke cell culture media for perfusion applications that can generate five-fold increases in volumetric productivity when compared to traditional fed batch/enriched fed batch media (Lyons, 2017) and the Modular Automated Sampling Technology (MAST) application which is capable of sterile sampling from a maximum of 10 bioreactors and transferring the acquired samples to a maximum of 4 analysers up to distances of 80 feet (Capsugel, 2016). Developments in sensor technologies involve improvements in the ruggedness and accuracy of electrochemical and optical sensors for the measurement of pH, temperature, DO and advances in capacitance and radio frequency impedance probes used for biomass characterisation. Process automation has seen development of ‘soft’ or software sensors. The sensors are comprised of a hardware sensor (e.g. Raman, NIR) coupled to a virtual sensor (microprocessor) which processes and models output from the hardware sensor to enable monitoring and/or controlling of manufacturing activity. Process control can be effected via either open loop or closed loop infrastructures. Real Time Release Testing (RTRT) will be enabled by this technology.

The technological advances listed above plus others demonstrate that CM is achievable using technologies that are currently available. However a major drawback with the technologies is the limited standardisation and interoperability between them. This results in the inability to operate in a ‘plug and play’ manufacturing environment and restricts manufacturers to employing a limited number of exclusive technology vendor(s) and necessitates a significant commitment of resources when switching between different vendors applications. This is a significant hurdle for CM which typically operates for extended periods of time and requires manufacturing equipment and machinery to operate precisely, accurately and reliably to assure production of product with appropriate quality, safety, potency and efficacy. Another challenge is the management and storage of the vast amounts of process data that will be generated. Industry is addressing the challenges, inter alia, through increased collaboration between manufacturers, technology vendors and regulators for example the Biophorum Operation Group (BPOG) technology roadmap initiative (2017) and ISPE GAMP and Good Engineering Practice Guides.
Cultural Attitude
Pharmaceutical manufacturing has operated in batch mode since the beginning of the industry. Product is manufactured in discrete, largely isolated, sequential steps with initial step outputs being used as input material for subsequent steps. This has created a culture where operations are performed in a largely independent manner with minimal interaction with other parts of the manufacturing train. Work practices and guidelines developed which reflected and reinforced this fragmented manufacturing environment. Examples include the discrete and separate functions of manufacturing, quality and information technology usually found in pharmaceutical manufacturing sites and the upstream and downstream processing functions in biopharmaceutical manufacturing sites. Regulatory guidelines also reflected the batch mode mentality for example process validation typically involved successful manufacture of three discrete batches of the drug with no subsequent assessment of the process necessary. Current regulatory thinking, as outlined in the ICH quality guidance documents, emphasises full process and drug product understanding (QBD, risk management etc.) and life-cycle verification approach to validation. This involves cross functional collaboration and information sharing with the focus on ongoing monitoring and verification of process performance. CM is the manufacturing manifestation of this mode of drug discovery, development and manufacture. It will require a fundamental change to current attitudes to manufacturing whereby individual functions (USP, DSP, manufacturing, quality, IT etc.) will need to collaborate more and work as a coherent and holistic team in manufacturing drug product. Work practices will require updating, manufacturing trains will be reconfigured (ballroom mode, modular units etc.) training and upskilling of staff will need to be provided (data analytics, model management and maintenance, data integrity) and more extensive collaboration with specialist SMEs (technology vendors, educational establishments) will be necessary. This will require extensive support, guidance and motivation from industry senior management, regulatory authorities and SMEs.
Conclusion
The current business environment for the pharmaceutical industry is exciting and challenging with change being the one constant factor. The immuno-therapy field is growing exponentially with the discovery of new modalities and indications fuelling growth and innovation in the biopharmaceutical sector. Small molecule development and manufacture is also experiencing significant change with an industry wide move to outsource this activity to CDMOs. The primary business drivers have been identified as flexibility, speed, quality and cost and CM will play a key role in transforming the industry to better support the market demands. Technology advances for, inter alia, sensor probes, automation software, perfusion, remote sampling and purification allied with regulatory encouragement and support have positioned CM to be the next big innovation within the industry. It will most likely be implemented with new drug product launches and will require a disruptive change to current work practices but the benefits and advantages vastly outnumber the challenges.
Glossary
BPOG : Biophorum Operations Group
CDER : Center for Drug Evaluation and Research
CDMO : Contract Development and Manufacturing Organisation
CM : Continuous Manufacturing
DO : Dissolved Oxygen
DCS : Distributed Control Systems
DSP : Down Stream Processing
FDA : Food and Drug Administration
GAMP : Good Automation Manufacturing Practice
ISPE : International Society of Pharmaceutical Engineering
MAST : Modular Automated Sampling Technology
NIR : Near Infra-Red
PAT : Process Analytical Technologies
QBD : Quality by Design
RTRT : Real Time Release Testing
RWD : Real World Data
SCADA : Supervisory Control and Data Acquisition
SME : Subject Matter Expert
SUB : Single Use Bioreactor
USP : Up-Stream Processing





