
Biosimilars, also known as follow-on biologics or similar biological medicinal product (henceforth referred to as biosimilars) are products that are similar to, but not the same as, an innovator’s drug. Since they are expected to cost less than an innovator’s drug, they bring affordable alternatives for treatment, for patients who previously could not afford them. Unlike generic drugs, biosimilars are generally regarded as therapeutically interchangeable.
From a historic perspective many biopharmaceutical companies make manufacturing related (process, raw material, equipment, process parameter etc.) changes throughout the product’s lifecycle. In case of biosimilars, it is well accepted that even small changes can potentially affect quality, safety and efficacy of the drug substance and the drug product.
Strategies of product development vary from company to company and from product to product. The approaches towards development and the extent of development vary, depending on the complexicity of the molecule. During development, understanding of product and process knowledge increases. This understanding is essential for determining all potential risk factors that impact product quality (CQAs) and control of these factors during the manufacture of drug substance and drug product becomes critical. In case of biosimilars, these risk factors are not only to be determined but also the process needs to be controlled in a way so as to match the product to RMP more closely.
The US Food and Drug Administration (FDA) offered a peek into its thinking on biosimilar drugs recently, by saying that because of their complexity, a “one size fits all” approach will not work. Biosimilar product should be developed, manufactured and controlled according to product specific pharmacopoeial monographs, country specific regulatory guidelines, ICH guidelines, WHO guidelines etc. An appropriate biosimilarity and characterisation plan is required to demonstrate that the biologic and RMP have similar profiles in terms of quality, safety and efficacy. The quality issues relevant for demonstration of comparability for similar biological medicinal products containing recombinant DNA-derived proteins are also addressed in the EMA’s “Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substances: quality issues”
Products which require high run rates, high success rates and minimal cost of goods (COGs) should have “full-blown” process characteriation efforts. Products with lower run and success rate requirements still require successful validation, but examining interactions and combinations of parameters at the edge of their prescribed range may add little value. Quality Attributes can be laid out in order of their Criticality to Product’s safety and Efficacy.
Manufacturing process of Biosimilars
During development state-of-the-art concepts must be considered (e.g. characterisation of cell bank, viral safety, robust purification, identification of excipients, formulation, degradation study, etc.). Process related and product related impurities also must be considered and it is important to demonstrate the consistency and robustness of the process in controlling these impurities.
As described in ICH-Q5E, comparability exercise should be considered when a change is introduced into the manufacturing process (either for active substance or drug product) during development or post approval. For the purposes of clarity, any comparability exercise for process changes introduced during development should be clearly identified and addressed separately from the comparability exercise done during product development vis-a-vis the reference medicinal product. The latter is mandatory for biosimilars.
Pros and Cons of Biosimilar Development Pathway for US Market

With an abbreviated biosimilar development pathway finally being formulated by US-FDA companies must decide whether to adopt the 351(k) route or forego it in favor of the traditional biologics license application (BLA) pathway. Companies will need to compare the advantage of getting to the market a few years faster and with much less development risk with the 351(k) route against potential negatives such as no market exclusivity, indication for use same as in the pivotal clinical phase-3 study, and extensive post-market surveillance.
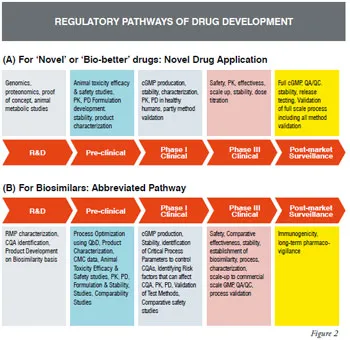
Given the intense competition in the rapidly evolving US market for biosimilars, the question often raised is - whether it is better to develop improved versions of the approved protein therapeutics (so-called “Biobetters”) taking the Novel Drug Application (NDA) route [Figure 2(A)] to get 12 years of market exclusivity against risking the “Abbreviated Biosimilar” developmental route [Figure 2(B)] with only 180 days exclusivity (for the first biosimilar) or no exclusivity? Another risk factor to consider is even after completion of critical early-stage studies for a biosimilar (i.e. CMC, nonclinical, clinical phase-1 / human safety, PK/PD) there is no guarantee that the product will be considered a true biosimilar (interchangeable with the innovator’s product).
Complexicity of Biopharmaceutical product
The development of a biosimilar product involves a stepwise approach of optimising the production process, characterisation of the product (physicochemical as well as biological) followed by pre-clinical and/or clinical studies. It may be noted that demonstration of similarity for a biosimilar product to the innovator product is a pre-requisite for the reduction of requirements for pre-clinical and clinical studies before gaining final marketing approval through an abbreviated regulatory process in most countries / regions. Identification of any significant differences in quality, safety and efficacy studies would mean the need for a more extensive pre-clinical and clinical evaluation and the product will not qualify as a biosimilar. Characterization study matrix are shown below in Figure-1
Biosimilar product needs to be studied in comparison with an approved product, referred to as the reference medicinal product (RMP), to establish similarity to the approved drug. The RMP provides the basis for dose selection and route of administration and is utilized in the comparability studies to support the application for regulatory approval. Comprehensive information on the RMP is therefore the basis for establishing safety, quality and effectiveness of biosimilars and to reduce requirement of pre-clinical and clinical data. An approved biosimilar drug cannot be considered as RMP, as the reference product should be the one that has been licensed on the basis of full quality, safety and efficacy data obtained from Complete Drug Development Pathway as shown in figure-2. Same RMP should be used throughout the development of a similar biologics i.e. manufacturing process, comparability exercise, pre-clinical and clinical evaluation.
RMP evaluation is critical in early stage of product development as it helps in setting the product specifications. Pharmacopoeia Product Monograph may or may not be available at the early stage of product development and hence RMP evaluation is key for successful biosimilar development. RMP evaluation has got an enormous power to refine your process so that you set the process parameters limits to achieve the desired biosimilar quality. Biosimilar developer must procure different lots of RMP at various stages of product development as innovators also conduct improvement of their process and there is a likelihood that the biosimilar under development may looks inferior at later stage of development, if the innovator improves the purity profile.
At times, the innovator makes significant changes in the characteristics of the approved product on the market (RMP), making it mandatory for the biosimilar developer to make changes in the process to restore comparability. Extensive structural, physicochemical, and biological methods should be applied to detect even “slight differences” in all relevant quality attributes affecting safety and efficacy. Analytical methods used should be appropriately qualified and validated with relevant information like use of standards and reference materials. Although the ability to fully characterise biosimilars have improved dramatically with improvements in analytical tools and techniques, there are no definitive technologies yet developed to predict immunogenicity of a biosimilar product, let alone determine changes in its immunogenic potential after making changes in the manufacturing process.
Challenges Versus Rewards
The field of biosimilars presents some significant challenges – safety, regulatory, legal and economic – which are still being discussed and debated in different forums. Most notable is the fact that, unlike the rather simple and straightforward process of introducing a generic equivalent of an original chemical (small molecule) drug, the process of introducing a biosimilar to an original biologic drug is far more complex and requires a battery of test (orthogonal methods) to characterizing the molecule as shown in figure-1. Consequently, generic drugs are sometimes referred to as “carbon copies” where as biosimilars are considered as ‘non-identical’ to the approved innovator drug.
Biotechnology and pharmaceutical companies have been watching the FDA’s decision process closely, as the market for biosimilars promises to be large. The global biosimilars market is expected to hit $19.4 billion by 2014, with a compound annual growth rate of 89.1 per cent from 2009 to 2014. Asia was the dominant market in 2008 because it commercialised products early, but the US is expected to dominate in 2014, spurred by the US market opening to the biosimilar products, according to a research analyst.








