
SBV Technology and Eradicating the Risk of Contamination in Aseptic Manufacturing
Christian Dunne, Global Product Manager ChargePoint Technology
Containment is key during aseptic processing and this has led to the development of new transfer methods that are both enclosed and minimise the requirement for operator intervention. In this article, the author discusses modern containment methods and the advent of split butterly valve technology to improve production processes..
Introduction
The global CMO Market is growing at a CAGR of 12–13 per cent between 2018 and 2022. The growth is attributed to increased outsourcing of generic drugs by Big Pharma companies. Additionally, medium and small pharma and biopharma companies, who do not possess adequate infrastructure, will also outsource, thereby driving the market. Presently, non-sterile drug manufacturing dominates the global CMO market. However, the sterile manufacturing market is expected to grow at a higher rate (15 per cent) than the non-sterile market (9 per cent) thru 2022.
Aseptic Processing
Manufacturing environments contain many potential sources of contamination which present significant hazards during the manufacturing of biopharma products. It’s important for firms to ensure successful containment to prevent endotoxins, microorganisms or particles entering the manufacturing environment as this could put patient safety at risk.
Equipment, materials, and people within the manufacturing environment can all offer potential sources of contamination if not suitably controlled. In esponse to this need we have seen multiple technologies starting to be developed, intended to allow the safe and sterile transfer of sterile APIs, biologics and biosimilars during processing. Isolators and Restricted Access Barrier Systems (RABS) have become commonplace technologies used to meet these needs in recent years.
Regardless, both of these solutions have their drawbacks. Isolators are typically challenging when it comes to transferring materials in and out of the chamber and this can cause productivity delays during the startup and shut off processes. Furthermore, RABS offer lower integrity chambers and this solution also requires sterilisation or manual cleaning processes such as Steaming in Place (SIP) between uses and this has increased time pressures which can cause delays.
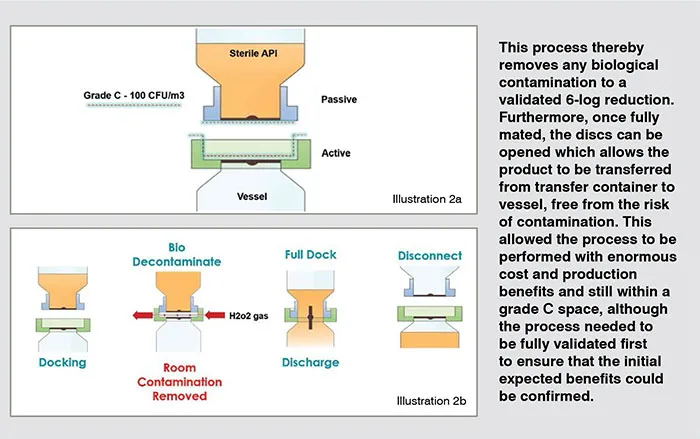
Aseptic SBV enables decontamination to take place in a closed environment. Once sealed, a gap is created between the two discs and Hydrogen Peroxide (H2O2) gas can then be flushed through this enclosure which decontaminates the space. Chemical Indicators (CIs) are used to validate and reassure the users that full coverage of the enclosure has been achieved. Biological Indicators (BIs) are then used to ensure a 99.9999 per cent, also known as a 6-log reduction in bacterial spores has been achieved.(Figure:1)

Case study
Sterile API Addition to a Mixing Vessel
A Contract Development and Manufacturing Organisation (CDMO) specialising in blow-fill-seal technology required a sterile solution for transferring drug substance during aseptic production. Its capabilities extend well beyond manufacturing, with an in-house development team specialising in all aspects of bringing a product to market - from lab scale batches, regulatory filings, scale-up, manufacturing and distribution.
Challenge
The CDMO was looking to solve the issue of charging sterile drug substance into a mixing tank. This is a common problem with all aseptically prepared products.
It was imperative to the process to sustain sterile conditions whilst docking a vessel to the container and then moving solid drug substance to form liquid. The product was sterile filtered as it was passed through the filler to become a completely dissolved liquid. However, in this application, the product being delivered to the filler was a suspension and so this was not a possibility.
The entire process needed to be performed under strict aseptic conditions, in order to remove the need to upgrade the whole room from a grade C cleanroom to grade A or introduce an over pressurised grade-A area around the point of fill.
Conventionally RABs and isolators would have been favoured in this situation due to the benefits which include enhanced sterility assurance. However, these both require a high initial and longterm capital investment and also require significant footprint within a facility so the company decided to search for a solution that was more appropriate to their application.
Application
An aseptic bio-valve product was selected to overcome these aforementioned shortfalls, providing a fully sealed powder transfer attached to the inlet port of the vessel. This valve can be pre-steam sterilised alongside the vessel, unlike traditional conventional connections incl SBVs (see illustration 1a/b). Once connected, it removes any contamination that might have come from the environmental surroundings thanks to the mating faces of the transfer being fully controlled and validated (see illustration 2a/b). (Figure:2) (Figure:3)


Validation
Firstly, microbiological validation must take place. This produces a validated decontamination cycle which enables the H2O2 gassing phase. Following this, there follows four phases which the generator will run to ensure that a fully validated gassing cycle is achieved every time.

Firstly, it’s important to condition the chamber to reduce the humidity and provide an ideal condition for biological kill. Following this dehumidifying step, the VHP is introduced to the chamber gradually for increased decontamination. The VHP concentration is then maintained in order to deactivate any microbiological activity within the chamber. Finally, once biological decontamination is completed, the VHP is removed from the system so that no harmful levels of residue are left. Typically, the acceptance level is 1ppm although in this instance 0.4ppm was the achieved level. The CDMO used a lower residue limit to ensure they had a robust system with no chance of contamination of the product due to gas residue.
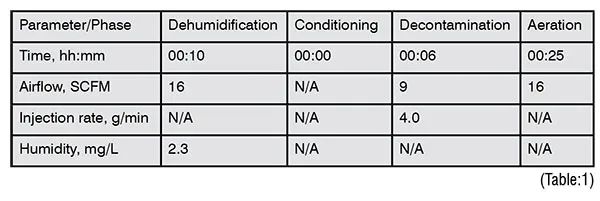
The complete decontamination cycle can take as little as four minutes. For this particular application, the process was only being performed once a day and to ensure a robust cycle was produced, additional time was added to each of the critical phases, ensuring that decontamination was confirmed, and gas was aerated from the system. This resulted in a 41minute full cycle. (Table:1)
Primary cycles employed Cis to control H2O2 distribution. When acceptable CI levels were achieved BIs were introduced to confirm full decontamination was attained. Once both the BIs and CIs were collected, their colour change was analysed to confirm that even vapor distribution has taken place. The BIs were then relocated to a Spordex culture media and placed in an incubator (between 55-60OC. They were then kept under observation for seven days and analysed for potential microbial growth).
Once the cycle was developed it was then performed three times to form the Performance Qualification (PQ) for this element of the process. Multiple media runs were performed prior to validation in order to fully validate the system and proving successful, these were then carried forward with three media runs at PQ. The sterile hold achieved greater than 10 days with product transferred to the container and with the bio valve positioned in the closed interlocked location. The sterile hold period was achieved for the passive section (product in transfer container) for 48 hours.
Conclusion
The installation is now fully operational. As well as benefiting from low capital equipment cost, a smaller footprint and improved installation, the CDMO can now boast improved sterility assurance, a greater ease of use for operators, and low maintenance which has certainly improved the CDMO’s process.
As the requirements of complex APIs continue to evolve, it will become increasingly important for technologies in the sector to be agile in order to adapt to new demands and changing customer needs.
Author Bio

Christian Dunne is the global product manager at ChargePoint Technology for the sterile containment solutions. For the past 20 years Christian has been creating innovative solutions for the pharmaceutical, biotech, cell therapy and fine chemical industries to overcome high potency containment and aseptic processing challenges. His technical expertise spans high containment isolators, grade A (ISO5) sampling & dispensing facilities, together with R&D and production filling line restricted access barrier systems (RABS) and isolators.








