Safety and efficacy of pharmaceuticals are two fundamental issues of importance in drug therapy. The safety of a drug is determined by its pharmacological-toxicological profile as well as the adverse effects caused by the impurities. The impurities in drugs often possess unwanted pharmacological-toxicological effects by which any benefit from their administration may be outweighed. It is quite obvious that the products intended for human consumption must be characterised as completely as possible. The quality and safety of a drug is generally assured by monitoring and controlling the impurities effectively. Thus, the analytical activities concerning impurities in drugs are among the most important issues in modern pharmaceutical analysis.
Regulatory Aspects
The International Conference on the Harmonisation of the Technical Requirements for Registration of Pharmaceuticals for Human use (ICH) which took place in Yokohama, Japan in 1995, has released new guidelines on impurities in new drug products. Analytical procedures should be able to separate all the impurities from each other and the method should be optimised to separate and quantify them in the dosage forms. Such methods are to be validated demonstrating the accuracy, precision, specificity, limit of detection, quantitation, linearity, range and interferences.
- The validation of analytical procedures i.e., the proof of its suitability for the intended purpose, is an important part of the registration application for a new drug.
- Additional peak tailing, peak resolution and analyte recoveries are important in case of chromatographic methods.
- ICH guidelines serve as a basis worldwide both for regulatory authorities and industries to bring the importance of a proper validation to the attention of all those involved in the process of submission of drug master files (DMF).
The analytical research and development units in the pharmaceutical industry are responsible for preparation and validation of test methods.
Origin of Impurities
- Last intermediate of synthesis
- Products of incomplete reactions
- Products of over reactions
- Impurities originating from starting materials
- Impurities originating from solvents
- Impurities originating from catalysts
- Products of side reactions
- Degradation products
- Enantiomeric impurities
- Residual solvents
- Inorganic impurities
- Impurities in excipients
Analytical Methodology
- The use of strict specifications to control drug quality is possible only when suitable analytical methodology exists.
- Before the advent of modern analytical separation techniques such as HPLC and GC, impurity levels of a few Per cen were considered acceptable.
- Separation methods are developed to detect and measure virtually any impurity, which is present at levels exceeding 0.1 per cent and often can be used at even lower levels of detection.
- For the last few years, the use of HPLC has gone from being a laboratory curiosity to the current state where HPLC is the workhorse of all analytical facilities.
In the early days of high performance liquid chromatography (HPLC), detection for qualitative or quantitative analyses often was carried out by collecting fractions and analysing them off-line using gravimetric or wet chemical techniques. It wasn't until the 1940s and 1950s that the first online detectors for liquid chromatography, the refractive index (RI), and conductivity detectors appeared on the scene, and while they were certainly an improvement over off-line approaches, neither detector was particularly sensitive. In the 1960s, the first ultraviolet (UV) detector for HPLC was introduced, and subsequent improvements in design led to better sensitivity and improvements such as variable wavelength and diode array UV detectors. While a truly universal HPLC detector with the kind of sensitivity achieved in GC–FID is still elusive, many different types of detectors have been developed since the early UV, RI, and conductivity detectors that have been very successful for a wide variety of general or specific HPLC applications.
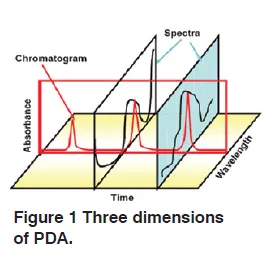
When Photodiode Array Detector (PDA) is combined with a separating column, three dimensions become available; wavelength, intensity and chromatographic retention. A particular substance appears as a point in this three dimentional space.
Peak Purity Function is useful to detect if a peak is only from one compound or if it contains coeluting peaks. It is done by comparing the similarity of the spectra at different points on the peak.
Advantages:
- PDA Detector could analyse a sample simultaneously at many different wavelengths.
- UV Visible spectra are useful for compound identification, checking peak purity, as well as finding the optimum absorbance for the compounds.
- UV Visible spectra of many compounds could be stored in the spectrum libraries, which are useful for compound identification.
- Relatively robust to temperature and flow rate fluctuations
- Compatible with gradient elution.
Disadvantages:
- Slightly less sensitive than UV-Visible detector.
- Detection of solutes that possess a significant chromophore.
Light-Scattering Detectors
Recent improvements in the ability to efficiently nebulise an HPLC column effluent has led to increased utility of light-scattering detectors. The most popular detection method of this type is evaporative light scattering detection (ELSD). ELSD works on the principle of evaporation (nebulisation) of the mobile phase followed by measurement of the light scattered by the resulting particles. The column effluent is nebulised in a stream of nitrogen (carrier gas) in a heated drift tube, and any nonvolatile particles are left suspended in the gas stream. Light scattered by the particles is detected by a photocell mounted at an angle to the incident light beam. Carrier gas flow rate and drift-tube temperature must be adjusted for whatever mobile phase that is used. Detector response is related to the absolute quantity of analyte present, and while decreased sensitivity will be obtained for volatile analytes, unlike the UV detector, no chromophores are required and it has orders of magnitude more response than the RI detector. ELSD also has the advantage over RI detection in that the response is independent of the solvent, so it can be used with gradients, and is not sensitive to temperature or flow rate fluctuations. Mobile phases of course must be volatile, similar to those used for MS detection. Linearity can be limited in some applications, but is certainly quantitative over a wide enough range if properly calibrated. Recent applications of ELSD also have been extended to UHPLC.
Charged-Aerosol Detection
Corona charged aerosol detection (CAD) is a unique technology gaining in its popularity in which the HPLC column eluent is first nebulised with a nitrogen -carrier gas to form droplets that are then dried to remove mobile phase, producing analyte particles [5,6]. The primary stream of analyte particles is met by a secondary stream of nitrogen that is positively charged as a result of having passed a high-voltage through platinum corona wire. The charge transfers diffusionally to the opposing stream of analyte particles, and is further transferred to a collector where it is measured by a highly sensitive electrometer, generating a signal in direct proportion to the quantity of analyte present.
Because the entire process involves particles and direct measurement of charge, CAD is highly sensitive, provides a consistent response, and has a broad dynamic range, which offers advantages when analysing compounds lacking UV chromophores. Often compared to other universal-type HPLC detection methods, like RI and ELSD, CAD has been shown to be much easier to use, and unlike RI, can accommodate gradients. In addition, CAD response is not dependent upon the chemical characteristics of the compounds of interest, but on the initial mass concentration of analyte in the droplets formed upon nebulisation, providing a much more uniform response as opposed to, for example, UV, where responses can vary dramatically according to the wavelength used and the extinction coefficient. Table I shows some common HPLC detector properties.
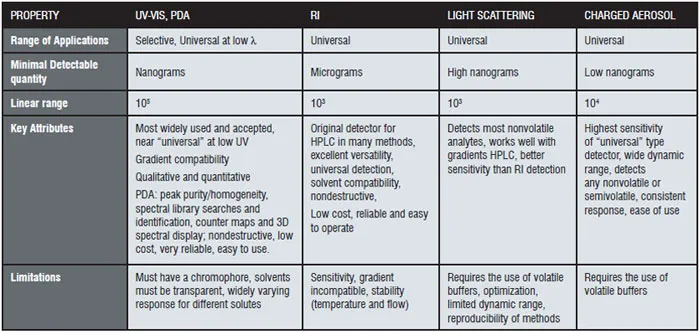
In CAD and in ELSD the effluent of a chromatographic system is nebulised, using a flow of nitrogen, and the resulting aerosol is transported through a drift tube where the volatile components and solvents are evaporated. In the last step in CAD the dried particle stream is charged with a secondary stream of nitrogen that has passed through a high-voltage platinum wire and the resulting charged particle flux is measured by an electrometer. In case of ELSD the signal is proportional to the number of photons scattered from the residual solid fraction that has been introduced into a detection cell. Operation is simple, requiring only setting of few controllable parameters, among them the gas input pressure, the temperature range and signal output range. Like ELSD, CAD system is a mass-dependent detector and the generated response does not depend on the spectral or physicochemical properties of the analyte as in a specific UV detector, which is a concentration-dependent detector. Theoretically this means that CAD and ELSD systems as bulk property detectors generate a similar response for identical amounts of different analytes. CAD generates a nearly constant response under isocratic conditions for compounds at similar concentrations. The relative magnitude of the CAD system response correlates with the relative mass of the analytes. The estimated relative response factors (RRFs) of known impurities were comparable with accurate values obtained from linearity data. It could be a fast, convenient, and accurate method to determine RRFs of known and unknown impurities. Important for accurate quatitation of impurities when pure standards cannot be obtained.
Small-particle columns: One area of significant advancement in drug substance impurity analysis is the utilisation of smaller, porous silica particles (<2>
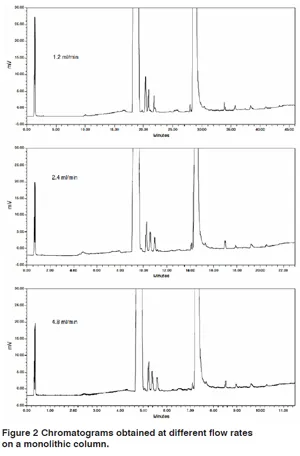
Reversed Phase-Monolithic silica columns have received significant attention for achieving faster analysis times using higher flow rates than are accessible with typical packed columns. Monolithic columns are prepared by polymerisation of monomers that result in a continuous porous rod rather than a packed bed of individual particles. This interconnected skeleton provides macro pores (2 µm) with low resistance to flow while mesopores (13 nm) provides high efficiency chromatographic performance. Advantages: Pressure increases for monolithic columns were modest and well within the operating ranges of conventional pumps even at flow rate up to 9 mL/min. Reduced run times at higher flow rates without loss of resolution (Figure 2). Wu et al found that the monolithics were not as retentive as packed columns and lower solvent strength were required for comparable retention. Good robustness and increased run times were observed for methods with intermediate flow rates. No significant column aging effects for monolithics were noted after 22,000 column volumes of use. Gerber et al also reported good separations with decreased analysis times and good column stability. Several additional applications: chiral separations, hydrophilic interaction chromatography, ion chromatography, capillary chromatography.
Stationary phase development: polar–embedded phases incorporate a polar group (e.g. amide, carbamate, urea or ether) into the alkyl chain. These phases are created to reduce silanol interaction with basic analytes. And found use as stable phases for highly aquous separations and in the intraction of polar analytes. Benefit of polar-embedded phases is the alternative selectivity afforded when compared to a standard alkyl phases (C18 or C8). Grumbach et al demonstrated that polar embedded phases are best for retention of polar analytes Mobile Phase pH: significant impact on the retention of acidic and basic analytes. The use of silica based phases above pH 7.5 is not feasible due to dissolution of the silica particles. Stable for pH 12: Zorbax extende (Agilent) –incorporates a bidentate bonding and double end-capping Xterra (waters), which incorporates a hybrid particle that is a combination of silica and organic (methyl silane) components.
Systematic or Automated Method Development
Method development for impurities in pharmaceuticals can require significant time and effort.
Method development HPLC systems- switching among multiple columns and mobile phases as well as optimising the separations based on the data obtained.
Softwares: Chromsword software (IRIS Technologies) to predict retention using a model derived from analyte structures and column properties. ACD/LC Simulator allows prediction of chromatographic separations based on structure and column chemistry. AMDS (Waters) allows the user to choose columns to screen and then optimises the separation on one column. DryLab (Rheodyne) LC-modeling software.
QbD Approach to Chromatographic Method Development:
1. Method objective
Analytical Target Profile (ATP) for measurement of impurities present in API.
Impurities controlled at 0.2 per cent with reporting threshold of 0.05 per cent.
ATP: The method should be able to quantify impurities in the presence of API, as well as any other potential impurities/degradation products over a range of 0.05-0.5 per cent relative to the API.
Combined accuracy and precision of the method must be within the range of 100 per cent ±10 per cent
2. Method design Phase
Suitability of different techniques such as :
LC/UV/MS (HPLC or UHPLC), GC-FID/MS, SFC-UV/MS
To meet specificity, sensitivity, accuracy and precision requirements
Capability for on/at-line measurements, analysis time and cost, sample preparation
Define starting conditions from which method evaluation and optimisation can be performed.
3. Method Evaluation Phase
Risk assessment of the method conditions in order to identify all potential sources of variation in the practice of the intended method. Such as column temperature, gradient profile, buffer concentration, flow rate, sample/standard preparation factors (sonication time/solution volumes), resolution between critical pairs, environmental factors: humidity, reagent source, analyst training-ruggedness assessment.
Inorganic Impurities
Impurities are classified (ICH Q3A guidance) as organic, inorganic, and residual solvents. Some inorganic impurities are toxic at low levels, and these impurities should be monitored to ensure safety. Sources of inorganic impurities include those that are deliberately added to the process (e.g. catalysts), undetected contaminants from starting materials or reagents, leaching from pipes and other equipment from naturally derived plant or mineral sources. The level of each inorganic impurity should not exceed the limit defined or otherwise specified in the individual monograph.
Equipment: One of the following plasma spectrometers is required for an analyst to perform this multi-element analysis:
- Inductively coupled plasma–atomic (optical) emission spectrometer.
- Inductively coupled plasma–mass spectrometer.
In addition, a closed-vessel microwave digestion system may be required for the preparation of test materials.
What is ICP-MS: Inductively Coupled Palsma-Mass Spectrometry
ICP-MS Elemental analysis with:
- Wide elemental coverage
- Very low detection limits
- Fast analysis times (all elements at once)
- Wide analytical working range (upto 9 orders)
- Simple spectra
- Isotopic information
- High matrix tolerance
- Viable alternative to ICP-OES or GF-AAS
ICP-MS can:
- Measure almost any element at ppt to ppm levels in almost any material
- Measure all elements in a single analysis
- Distinguish different element species (speciation)
Main requirements for pharmaceutical analysis are:
- High sensitivity
- Good matrix tolerance
- Low levels of interferences
- Ease of coupling to separation techniques (CE, IC, LC and GC) for speciation analysis.
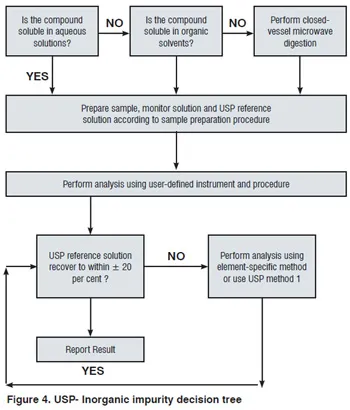
USP Reference Standards—
- USP Inorganic Impurities Class 1 Reference Standard for test articles soluble in aqueous solutions.
- USP Inorganic Impurities Class 2 Reference Standard for test articles soluble in organic solvents.
- USP Inorganic Impurities Class 3 Reference Standard for closed-vessel microwave digestions.
Conclusions
A major trend in LC for the determination of drug impurities is clearly towards faster separations with comparable or improved separation capability. This is indicated by applications of monolithic columns and short, sub-2-µm columns that may be operated at high pressures. Faster analysis times facilitate more efficient method development and exploration of a wide range of mobile phase pH, organic modifier and stationary phase in an automated fashion. Stationary phase development has focused on columns that provide greater retention for polar analytes, columns that will tolerate operation at pH values above 8 or columns that provide unique selectivity. Advances in detection include the CAD, which is likely to see increased application for non-UV-absorbing compounds.
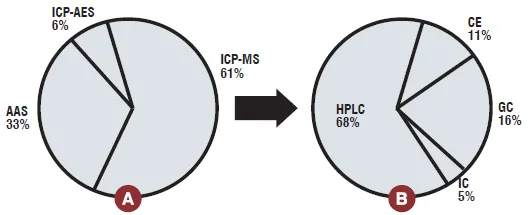
The ICP-MS is as an effective tool in impurity profiling of single, multi and speciation analysis of different elements present in bulk drugs and formulations. The particular advantage of ICP-MS, when compared to other techniques is speciation studies for many elements and separation ability of chromatography coupled to ICP-MS offers a versatile tool for speciation. HPLC was the most widely used chromatographic technique, which occupied nearly 70 per cent of speciation analysis. The use of ICP-MS for elemental specific assays for accurate determination of pharmaceuticals and impurities without the need for standards was also reported. Collectively, these advances will lead to improved capability for drug impurity profiling.










