
In the past three decades, biotechnology-led medicines have revolutionised the treatment of several life threatening and rare diseases. The substances produced by living cells and used in the treatment, diagnosis or prevention of diseases are referred to as biologic drugs or biologics or biopharmaceuticals or recombinant therapeutics. Since the approval of first biopharmaceutical, recombinant insulin in 1982, the range and market of biopharmaceuticals has grown significantly. More than 250 biological products approved in various countries and 500 new products are in pipeline. Biopharmaceuticals are now important therapeutic option for a variety of chronic and non-chronic diseases including the rare diseases.
Biologics are inherently different from chemicals drugs in terms of their source, structural complexity, fragility of the active substance, manufacturing, quality control and stability. Since chemical drugs have well-defined structure, their quality can be easily optimised and maintained, while the large and complex bio-molecules are difficult to manufacture everytime in a similar fashion and therefore, highly susceptible to heterogeneity. As a result, avoiding the batch to batch variation during their manufacturing is a highly challenging task. Besides, these manufacturing variations more often lead to immunogenic reactions and compromise the patient’s safety.
The patents of the first generation of biotechnology products have either expired or will expire shortly, thereby opening the market for introducing the follow on substitutes of the original biologics, these follow-on substitutes are popularly known as ‘biosimilars’. There are a number of terms used to describe these substitutes such as biosimilars, biologics, biogenerics, biopharmaceuticals, follow-on-proteins etc. These terms are confusing and do not include vaccines and blood products produced through recombinant route. Recently, Review Committee on Genetic Manipulation (RCGM), Department of Biotechnology has adopted a substitute term “Similar Biologics”, which is defined as recombinant biologics similar to the original innovator product based on the comparability studies.
In 2009, recombinant proteins accounted for more than 65 per cent of the total global biopharmaceutical sales. Growth in this class is expected to be low in most of the developed markets as a result of biosimilar entry and increasing cost containment measures. The market share of emerging markets (Brazil, Russia, India, China, Mexico, Turkey and South Korea) is likely to increase from less than 5 per cent in 2009 to more than 8 per cent by 2015. The similar biologics have the potential to provide affordable biotech medicines, however, the issues and challenges associated with them needs establishing the appropriate regulatory pathways to ensure quality, safety and efficacy. International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) was the first to recommend the guidelines for biosimilars but it is the European Union (EU), who implemented them first in 2006. EU was followed by several other countries like Australia, Canada, Malaysia, Japan and organisation like World Health Organization (WHO).

International status
European Medicines Agency i.e. EMA was first to develop a regulatory pathway for biosimilars, which they designated as ‘Similar Medicinal Biological Products (SMBP)’. Article 10(4) of the EU’s Code for Human Medicines Directive (2001/83/EC) was amended in 2004 (by Directive 2004/27/EC) to authorise the abbreviated approval of biologic products that claim to be similar to an original innovator product. However, the legislation leaves a wide margin of discretion to the Committee for Medicinal Products for Human Use (CHMP; EMA) to develop product class-specific guidelines that determine the extent of nonclinical and clinical testing required to establish the safety and efficacy of a SMBP. EMA first released overarching guidelines on quality issues of SMBPs containing biotechnology derived proteins as active substance followed by separate guidelines focused on non-clinical and clinical issues. In addition to these general guidelines they also drafted customized guidelines for different biotechnology based products such as Insulin, Erythropoietin (EPO), Granulocyte Colony Stimulating Factor (GCSF), Interferons and Growth Hormone.
In the USA, biologics are authorised for marketing through two regulatory pathways i.e. Biological License Application (BLA) procedure under Public Health Service Act (PHS, 1944) for regulation of biological produced by biotechnological methods (e.g., MAbs and therapeutic proteins) and abbreviated New Drug Application (NDA) procedure under Food Drug & Cosmetic Act (FD&C, 1938), which covers conventional pharmaceutical and certain natural proteins (e.g. insulins and growth hormone). The ANDA procedure allows approving only a limited range of biosimilars. In March 2010, US Federal Government amended Section 351 of the Public Health Services Act to create abbreviated biologic approval pathway for a “highly similar” biologic product. A biosimilar must possess no clinically meaningful differences in the safety, purity and potency from the original innovator product and the product may be interchangeable if the product is biosimilar and show no clinically significant difference to the reference product.

The new act also includes a 12 year data exclusivity period for all original products (with a six-month extension for products supported by paediatric studies). The act also provides one year data exclusivity to a first biosimilar approved for marketing to boost the availability of biosimilars. Though there is a mechanism now, the FDA is still not ready to give final shape to its biosimilar guidelines as a result of highly debatable data exclusivity and patent issues. Currently, both EMA and FDA guidelines are under revision.
WHO has developed a framework of general principles as monograph and part of its “Biological Standardization Process” and prepared a “Guidelines on evaluation of similar biotherapeutic products (SBPs)”. Biosimilar is designated here as “Similar Biotherapeutic Product (SBPs) and defined as a “biotherapeutic product claimed to be similar in terms of quality, safety and efficacy to an already licensed reference biotherapeutic product (RBP), which must have been licensed by national regulatory authorities on the basis of a full registration dossier”. However, WHO emphasises that its framework is generalised and will only apply to well-established biologics. Its guidelines emphasise the high standard of safety and require at least one clinical study for approval. In addition to pre- marketing safety study, the applicant has to submit a well devised plan for post-marketing clinical safety and surveillance.
International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) is a joint initiative involving both regulators and research-based industry representatives of the European Union, Japan and the USA in scientific and technical discussions of the testing procedures required to assess and ensure the quality, safety and efficacy of medicines. The goal of ICH is to promote international harmonisation by bringing together representatives from the three ICH regions (EU, Japan and USA) to discuss and establish common guidelines. In July, 1997, ICH recommended the harmonized tripartite guidelines, for the preclinical safety evaluation of biotechnology-derived pharmaceuticals. The active substance include proteins and peptides, their derivatives and products of which they are components; they could be derived from cell cultures or produced using recombinant DNA technology including production by transgenic plants and animals. The guidelines may also be applicable including recombinant DNA protein vaccines, chemically synthesized peptides, plasma derived products, endogenous proteins extracted from human tissue and oligonucleotide drugs. The primary goals of preclinical safety evaluation are: 1) to identify an initial safe dose and subsequent dose escalation schemes in humans; 2) to identify potential target organs for toxicity and for the study of whether such toxicity is reversible; and 3) to identify safety parameters for clinical monitoring. In these guidelines, the biosimilars have not assigned any separate name and the list of products covered is too wide though they are considering them on case-by-case basis and don’t consider them as ‘same’.
Indian scenario
In India, Review Committee on Genetic Manipulation (RCGM), Department of Biotechnology is responsible for preclinical, export and import approval of biotechnology based recombinant drugs, while Central Drugs Standard Control Organization (CDSCO) and the Drugs Controller General of India (DCGI) is responsible for approvals of clinical trials, new drug applications, marketing and the import of drugs in association with Directorate General of Foreign Trade (DGFT). The state drug control authorities are responsible for licensing a drug maker’s research and manufacturing facilities, while Institutional Biosafety Committees (IBSC) oversees the containment facilities.
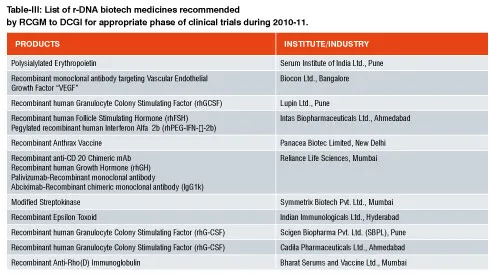
As of now, no separate pathway for approval of recombinant drugs under the category of “similar biologics” exists in India. Over 40 biologics are marketed in India, out of which around 25 are similar biologics. Another 25 similar biologics are in the final stages of development. In India, whichever similar biologics is available, they are approved as ‘new drug’ under Drugs and Cosmetic Act, 1940. Table-I represent the r-DNA technology based biopharmaceuticals already having marketing authorization. Table II & III includes the r-DNA technology based biopharmaceuticals for pre-clinical studies or recommended for clinical studies to DCGI by Department of Biotechnology in a financial year 2010-2011. Under the current relaxed regulatory environment, Phase I & II clinical trials are typically not required for similar biologics approval in India unless it is found necessary in special cases. Phase III trials with a minimum of 100 patients are mandatory for establishing bioequivalence.
Currently, there are two serious concerns about the Indian regulatory system related to similar biologics. First, number of drug regulatory authorities involved in approval procedure, which makes the overall process time consuming and unnecessary lengthy. Second is the lack of clearly defined guidelines for similar biologics approval. The Indian government acknowledged the need for tighter regulatory standards. The major regulatory initiatives have been taken to make the approval process streamline through establishing a single window mechanism. Department of Biotechnology (DBT) has drafted a bill for the development of Biotechnology Regulatory Authority of India (BRAI), which will be tabled in the monsoon season (2011) of Parliament for discussion and hopefully will be cleared for developing the BRAI as an independent authority. BRAI will be an “autonomous and professionally led body to provide a single window mechanism for the biosafety clearance of genetically modified products and processes.” In other words, BRAI will replace many of the other bureaucracies. BRAI will also include a training center for its biotech regulators, to build and maintain their professional competence.DBT is also in the process of preparation of separate guidelines for preclinical evaluation of similar biologics. DBT holds series of public meetings especially with Indian biotech industries to find out the issues related to the manufacturing and marketing of similar biologics. RCGM is a group of experts having both academic and on-field experience is entrusted for the formulation of similar biologics draft guidelines. RCGM is performing the judicious analysis of all the factors that affects the quality, safety and efficacy of a similar biologics product. It is pertinent to discuss some of these issues briefly.
A similar biologic is generally compared with an original innovator product to establish the safety and efficacy of the product. There are two major concerns related to the selection of a reference product. First, in the absence of availability of original innovator product in India, whether a similar biologics, already authorised for marketing in India, can be used as a reference product? Second, whether some of supporting data of a reference product may be used as a part of subsequent similar biologics clearance and approval? In general, the EMA Guidelines (2004) indicate that the chosen reference product must be authorised in the country on the basis of a complete dossier. Utilising the reference product data for similar biologic approval makes the process less time consuming and expensive but it is not acceptable on grounds of manufacturing heterogeneity often observed in biopharmaceuticals. Also any differences observed between a similar biologics and a reference product will have to be justified by appropriate studies on a case-by-case basis.
In general, the biophramceuticals are large, complex and heterogeneous molecules with more variable molecular weights in comparison to chemically originated small-molecules. The inherent complexity of these molecules makes their manufacturing susceptible to variation. Therefore, it is essential to maintain rigorous quality control at each step of manufacturing i.e. from fermentation to packaging. Changes may occur in the expression systems used for production, culture conditions (e.g. temperature and nutrients), purification and processing, formulation, storage and packaging. Small changes in, or differences occurred during manufacturing processes may have a significant impact on the quality, purity, biological characteristics and clinical efficacy of the final product e.g. “Valtropin”, a similar biologic growth hormone is different from its reference product “Humatrope”, probably because different cell lines are used in the production of these two drugs: yeast for Valtropin and Escherichia coli for Humatrope. Another example is the occurrence of pure red blood cell aplasia as a result of production of neutralising antibodies against a particular form of Epoetin-alpha.
Structural differences between proteins may arise for a number of reasons, including oligomerization, modification of the protein primary sequence, glycosylation patterns or the conformational state. The analytical homogeneity is extremely crucial between a similar biologic and reference product to maintain the quality of a product. There are numerous examples where presence of process and product related impurities resulted in substantial variation in a similar biologic. For example, almost 60 per cent of patients treated with recombinant human Growth Hormone (rhGH) powder manufactured by API Covance (Somatropin Sandoz powder) developed anti-GH antibodies and all patients developed antibodies against E.coli proteins, while only about 2% and 0% of patients develop anti-GH and anti-HCP antibodies respectively when treated with a innovator/reference product ‘Genotropin’ (Pfizer), [4]. The additional purification steps are included in order to ensure the tolerability and efficacy of the product.
Another hurdle in quality assurance is the inherent limitations of the biological assays. Therefore, the biological assays should be performed simultaneously and parallelly on a similar biologic and reference product under the same laboratory conditions to mitigate or reduce the assay errors. The most important among all the biological assays is the toxicity studies including toxicokinetic measurements such as determination of antibody titres, cross-reactivity and their neutralising capacity. A biological product may be toxic due to its degradation during storage in the distribution chain before reaching to the end consumer. Therefore, the comparability studies may also include the comparative data for accelerated and long term stability to make sure the quality and safety of a product during storage.
The analytical comparison of a similar biologic with a reference product does not guarantee about the behaviour of a product in human biological system. Therefore, it is undoubtedly essential that the applicant must submit elaborate in-vitro and in-vivo pharmacological and toxicological studies data as a part of application dossier. Safety pharmacology, reproduction toxicity, mutagenicity and carcinogenicity studies may not be required unless specifically desired in a particular case.
Generally, the biological products obtained from one system are immunogenic when introduced in another biological system. Mostly similar biologics are recombinant proteins and antibodies, their introduction might trigger a severe immunogenic reaction especially when the patient is already exposed to a molecule. Immunogenicity should be evaluated using appropriate studies and methods to characterize type, concentration and titre of antibodies. When neutralizing antibodies are detected, the impact on Pharmacokinetic (PK) and Pharmacodynamic (PD) parameters and overall efficacy and safety should be analysed well. This is particularly true for proteins with post-translational modifications such as glycosylation where small differences in glycosylation pattern can result in significant differences in immunogenicity profile.
Conventional generics of chemical origin are considered to be therapeutically equivalent to a reference product if it has pharmaceutical equivalence (i.e. identical active substance) and bioequivalence (i.e. comparable pharmacokinetics). The inherent complexity of biopharmaceuticals makes it difficult to avoid heterogeneity among different batches and from different manufacturers. The challenging task is to decide whether abbreviated clinical trial should be made mandatory or data requirement should be restricted and desired only on case by case basis. Clinical trial for a similar biologic is estimated to cost US$ 26.5-$53mn. After including the cost of approval through the regulatory process and the cost of marketing and detailing, the estimated cost would be US$ 50mn and US$ 300mn with a manufacturing plant. This is certainly a huge investment for any single copy-cat drug. The estimated cost will definitely reduce the profit margin. Therefore, the major question is can clinical trials be skipped by taking the comparability studies as standard in cases which are known to be safer. The argument in favor of restricting the clinical trials is that if a similar biologic is substantially similar to a reference product during comparability studies and then most probably it will behave in the same fashion in human system as the reference product. Argument in favor of making the clinical trials mandatory is that even animal (pre-clinical) studies do not guarantee the absolute safety and efficacy of any biological product in a human system. So, RCGM need to weigh the pros and cons of all possibilities related to clinical trials and then appropriately implement the best possibility in case of similar biologics.
Once a product is authoried for marketing, it is desirable to access and monitor its effect in terms of safety and toxicity in human population. In case, the Indian regulatory authorities restricting the clinical trials during approval of similar biologics, the post-marketing surveillance will be extremely important e.g. In Korea, three biosimilars of Epoetin-alfa i.e. ‘Eporon’ (Dong-A Pharmaceutical Company Ltd), ‘Espogen’ (LG Life Sciences), ‘Epokine’ (CJ Corporation) have been shown to differ in the activity, concentration, isoforms, structural stability from the reference product ‘Epogen’ (Amgen, USA) [5]. Like EMA, the Indian regulatory authority may ask companies to submit self-executable pharmacovigilance plan as a part of approval process or collect the similar biologics field samples randomly and then testing them in governmental laboratories. The second option is more practical especially in the absence of a very well established pharamcovigilance system in India. Recently, Central Drugs Standard Control Organization (CDSCO) has taken an initiative to establish a Pharamcovigilance Progarmme of India (PvPI), which is expected to be fully operational by 2015. PvPI will definitely open the door for the regulatory authorities to keep stringent on-field vigil for ensuring the safety and efficacy of drugs in general.
Conclusion
The huge Indian market and the margin associated with similar biologics is highly lucrative for many big players in the biotech sector. Many of them are very curious about the regulatory pathway not only in India but also in other markets like USA. Very soon, more biotech based drugs are going to be out-of-patent and the related guidelines will decide the strategy of investment, molecule selection and marketing plans. While framing the guidelines for similar biologics, Indian regulatory authorities will try to keep a balance between making available the high quality similar biologics at a cheaper price to Indian consumers and cost advantage to biotech industries so that they remain interested in manufacturing them. DBT has advocated to keep biotech medicines out of price control (DPCO) to facilitate growth of this industry for the benefit of society and has the aim that let market forces decide the price through competitive process.
References
- Global Biopharmaceutical Market Report (2010-2015) by IMARC available at http://www.imarcgroup.com/global-biopharmaceutical-market-report-2010-2015/ (Last accessed on 16-08-2011)
- Peterkova, V. et al. (2007). A Randomized, Double-Blind Study to Assess the Efficacy and Safety of Valtropin, a Biosimilar Growth Hormone, in Children with Growth Hormone Deficiency. Horm. Res. 68(6), 288-293
- Casadevall N, Rossert J. (2005). Importance of biologic follow-ons: experience with EPO. Best Pract. Res. Clin. Haematol. 18, 381-387
- Declerck et al. (2010). Biosimilars-Controversies as illustrated by rhGH. Curr. Med. Res. & Opin. 26, 1219-1229
- Deechongkit et al. (2006). Biophysical comparability of the same protein from different manufacturers: a case study using Epoetin alfa from Epogen and Eprex. J. Pharm. Sci. 95, 1931-1943
- Note: The views expressed are personal to the authors and have no relation with their official position in Department of Biotechnology.





