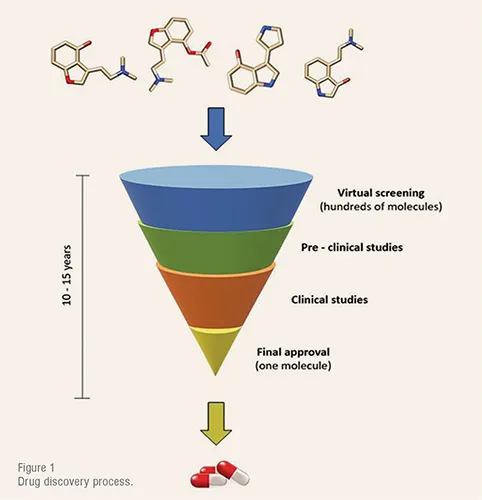
Drug discovery is one of the most time- and cost-consuming processes in the industry. From active pharmaceutical ingredients (API) discovery to final approval it involves a total duration of 10 – 15 years (Figure 1) and US$1 – US$2 billion investment.
After the protein target identification, the search for a specific drug-like molecule that matches the binding site is an arduous process which requires the screening of hundreds of molecules to test their bioactivity. Employing an experimental trial-and-error procedure, this task would take years to be accomplished. Thus, during the past few decades, different theoretical approaches based on quantum and molecular mechanics have been developed to simulate and test real systems before entering the laboratory.
The explosion of biotechnology resources has enabled the disease process to be better understood and evaluated for drug targets. So, nowadays, drug design cannot be imagined without computational aided methodologies such as CADD. Therefore, CADD has gained importance in the pharmaceutical industry, at research centres, and universities.
CADD involves techniques such as molecular docking or virtual screening, which are employed to inspect the binding poses of a ligand (or a database of ligands) on a particular receptor. Despite being much more efficient than the aforementioned experimental ones by far, these procedures require large calculations and extensive chemical data repositories to simulate their highly complicated systems. Hence, its related computational cost, in terms of memory and processing capacity, is huge.
Accordingly, to transcend computing barriers of the traditional personal computers (PCs), high-performance computing systems have been developed.
High Performance Computing (HPC) is the application typically used for solving advanced computational problems that are too large for standard computers and performing research activities through computer modeling, simulation and analysis.
Computer-aided drug discovery methodologies have become essential components of the drug discovery and Virtual Screening (VS) methodologies have emerged as efficient alternatives for the discovery of new drug candidates.
Protein-ligand interactions are common targets for medicine. Refining computational models would help in developing new medicines more efficiently. The problem of finding potential dual target ligands is addressed from different points-of-view: similarity analyses, QSAR-derived (Quantitative Structure Activity Relationship) ligandbased virtual screening studies, and molecular docking approaches.
Virtual screening (VS) refers to a group of in silico techniques oriented towards finding novel hit and lead compounds. The technique has become prominent in the last few decades as a means to study diseases such as malaria, diabetes and Parkinson’s disease. Although its computational cost is considerable, VS is one of the most used methods when it comes to assessing large chemical spaces and shorten the time devoted to in vivo experimentation. VS techniques are manifold and can be divided into two broad categories: structure-based,(SBVS) when the 3D structure of the target is known, and ligand-based (LBVS) when it is not. Alternately, LBVS depends on the notion of small molecules that connect the target and it comprises several methods, such as pharmacophore modelling, similarity searching and QSAR. The idea behind pharmacophore modeling is the alignment of two or more molecules to identify the pharmacophore features they share. Molecular similarity is then characterised by compounds with pharmacophoric features that resemble the identified pattern. Similarity searching is the preferred method due to its simplicity, which also makes it cost-efficient. It rests on the comparison of built fingerprints for molecules that can be easily compared while maintaining the information necessary to determine similar biological activity.
Evolution of Drug Discovery Software
Before the explosion of web technologies and the Internet, software packages used to be installed on local machines and users required physical or remote access to them. That was the way that Autodock, DUD and many other packages were used far in the past. This classic approach presents several disadvantages and limitations. First, users must be granted to access the machine where the software is. This approach may work well with few users, but it does not scale when the number of users increases. Moreover, it represents an important security issue because users should only be granted to run the software they require. Second, running a command-line programme implies an in-depth knowledge of the parameters in order to receive the expected result back. Unfortunately, drug discovery is a multidisciplinary science and not every user is familiar with all the concepts involved or has advanced computing skills as to deal with complicated options. Finally, installing a programme in a new machine might not be an easy task due to software dependencies. The destination environment must be compatible with the source one and provide all required dependencies. Once more, not every user is able to cope with certain steps of the installation process.
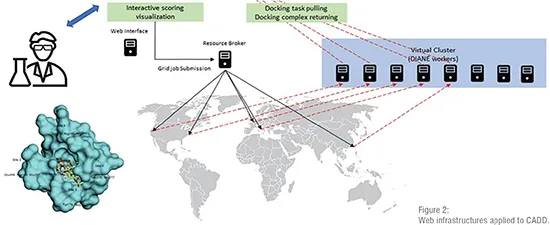
To overcome these drawbacks, web technologies came to the rescue. They provide a common, accessible and simple environment that hides the complexities of the underlying software and centralise the access to resources. By means of web servers, millions of users can execute drug discovery tasks at no installation cost. The advantages of web technologies in the context of drug discovery are several. As stated, they provide unlimited access to the published resources at any time and from any place (Figure 2). Security risks can be easily handled in one single entry point. Furthermore, only one installation is necessary, and it is transparent to users. In addition, since users are unaware of the hardware behind the web interface, it is simple to rely on an HPC infrastructure to carry out complex calculations and speed up time consuming tasks. Finally, the way the users interact with the software can be simplified through a nice web interface by limiting the number of input parameters and formatting the output to be more understandable. In the context of drug discovery several applications have moved to web environments, making it one of the main reasons behind the most important achievements experienced by CADD.
Nowadays, web servers are just the tip of the iceberg of the complex HPC infrastructure based on distributed computing environments such as grid computing, cloud computing and general-purpose computing on graphics processing units (GPGPUs). Biomedicine was one of the first areas that moved to distributed infrastructures, which rely on the idea that any simulation can be carried out by a service. In this type of paradigm supercomputers, clusters or workstations are services, but also databases and authentication mechanisms represent services. These new paradigms have the ability to execute multiple calculations simultaneously to accelerate the most time-consuming tasks resulting in a greater number of simulations and, consequently, in more chances to shorten the delivery time of new drugs.
Examples of web servers and databases
As discussed above, the use of web servers to deliver drug discovery services has gained a lot of importance in the recent decades. The main techniques, such as virtual screening, homology modelling or target prediction, have been migrated progressively to the web. Moreover, the data that those services consume have been published on databases that are accessible from the Internet.
The typical example of drug discovery process on the web are VS methods (e.g. similarity searching, molecular docking, pharmacophore modelling, QSAR). Due to their ability to deal with the overwhelming amount of data available in the chemical databases and the speed of calculation they exhibit, LBVS methods are suitable to be implemented on the web and many servers can be presented as a model. A typical example is SwissSimilarity1 which provides a simple interface to screen a disparate of datasets by a collection of 2D and 3D software packages. Another example is BRUSELAS server2 that carries out either similarity searching or pharmacophore screening tasks by combining different programs. It applies consensus scoring functions to return unbiased predictions and the results can be exported as a PyMOL session. Other similar servers such as USR-VS3 or ZincPharmer4 are more restrictive and only allow performing one type of VS on a single certain dataset but are also suitable in some cases. In general, all LBVS servers share some common features including the set of parameters (e.g. databases, software), simple and intuitive interfaces and the way the results are interpreted.
Many other techniques can also be executed via web servers. SBVS methods are represented by HADDOCK5, DOCK Blaster6, SwissDock7 or Blind Docking Server8 which implement different versions of molecular docking. 3-D QSAR, E-Dragon and MOLFEAT are web servers to perform QSAR related processes, including QSAR modelling and molecular descriptors calculation. Molecular dynamics (MD) is another remarkable approach in the context of CADD that has experienced the transition from command-line programs to the web. MDWeb, the Gromacs server, Vienna-PTM and MoSGrid are some representative examples of MD services on the web.
The evolution of HPC paradigms has opened a way to accomplish exhaustive investigations on contexts that were inconceivable a few decades ago. For example, the Cancer Genome Atlas helps to split genome data into small pieces for parallel processing. The Collaborative Genomic Data Model (CGDM) improves the performance of the queries on genomic databases. Rosetta@home is a distributed computing project that has been applied to research on Malaria and Alzheimer’s disease.
The aforementioned examples are only a sample of the full catalogue of CADD related services available on the web. Nowadays, almost every step in the drug discovery pipeline is automated and several web services provide that functionality through a nice interface. A complete collection of those services is maintained by the Swiss Institute of Bioinformatics and available at the Click2Drug portal9).
Though services are a key point in the drug discovery process, they are only half of the equation. The other half is made of data. Those data are stored in huge databases that frequently provide a simple web interface to browse them and, sometimes, even a web API to retrieve information through web services. The diversity of chemical databases that can be explored on the web is enormous, from the commercial ones such as Specs, Mcule, and May bridge to the freely accessible ones, such as PubChem, ChEMBL, ZINC, and GDB-17. They all contain many different attributes (e.g. identifiers, chemical descriptors, biological pathways) and records. Although they do not share a common structure, their web interfaces facilitate the navigation between them. In any case, current chemical databases can be easily explored via web browsers that allow running fast queries without having direct access to the physical store.
Conclusion
The development of web technologies has dramatically changed the drug discovery process and has paved the way to the most remarkable achievements in this area. Old fashioned command-line programs are now accessible by millions of users worldwide through simple web interfaces that hide all the complexities of the underlying software and encourage the use of HPC platforms to accelerate calculations. Simplicity in the use of software is a critical point in drug discovery because it is a multidisciplinary process and computing is only a part of it.
Nowadays, researchers can access a disparate set of web tools that implement different approaches for each task in the drug discovery process, ranging from single and well-defined steps (e.g. molecular descriptors calculation, prediction of ADMET properties) to more complex simulations (e.g. molecular dynamics, homology modelling) and calculations (e.g. molecular docking, QSAR). Furthermore, web development has changed data availability. Chemical databases are often freely accessible and non-expert users can explore them and build complex queries in their web browsers.
The application of these tools has allowed significant progress in the field of drug discovery, resulting in a shorter time to deliver new drugs, which has a great impact on society, especially in global emergency situations, such as Zika and Wuhan virus crisis.
References:
1 http://www.swisssimilarity.ch
2 http://bio-hpc.ucam.edu/Bruselas
3 http://usr.marseille.inserm.fr
4 http://zincpharmer.csb.pitt.edu
5 https://haddock.science.uu.nl
6 https://blaster.docking.org
7 http://www.swissdock.ch
8 https://bio-hpc.ucam.edu/achilles
9 http://www.click2drug.org












