
Since it was introduced in 1995, the Biopharmaceutics Classification System (BCS) has had a major impact on the regulation of oral drug products worldwide. Fundamentally, the BCS is a scientific framework for classifying drug substances (i.e. active ingredients) based on the factors that determine the rate and extent of absorption from immediate release (IR) solid oral dosage forms for the purpose of establishing equivalence in quality between test and reference drug products. By providing a basis for avoiding unnecessary in vivo studies, BCS helps significantly reduce the cost and time of developing drug products. BCS has gained importance worldwide as a drug product regulation tool; the system has been formally adopted by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), and the World Health Organization (WHO) (with some modifications) as a means to establish technical standards for waiving bioavailability (BA) and bioequivalence (BE) testing requirements for oral drugs. What role BCS should play in the consideration of biowaivers has garnered the attention of regulatory authorities in recent years. International differences in BA/BE-related guidelines should be reconciled in order to realise the potential cost benefits and time savings of the Biopharmaceutics Classification System.

How does the Biopharmaceutics Classification System work?
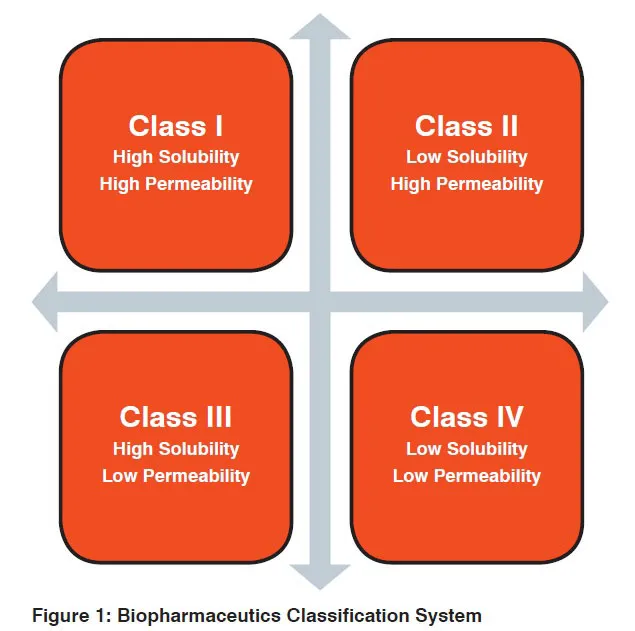
BCS is used to establish equivalence in applications for brand and generic medicinal products, variations, fixed combinations, extensions, and hybrids. Developed by Amidon and colleagues, the system classifies IR solid oral dosage forms on the basis of solubility and permeability parameters (when combined with dissolution testing). According to this schema, drug substances are categorised as having either rapid or slow in vitro dissolution and then classified based on aqueous solubility and intestinal permeability of the active pharmaceutical ingredient (API):
Amidon posited that human outcomes can be accurately determined based on the evaluation of two intrinsic properties of the API (permeability and solubility) and one property of the drug product (dissolution). The evaluation of these properties can be performed in vitro, therefore avoiding expensive and time-consuming testing in humans.
How is the Biopharmaceutics Classification System used?
BCS is being implemented by regulatory agencies around the world to predict the in vivo pharmacokinetic performance of drugs based on dissolution, solubility, and permeability measurements rather than on traditional BA and BE testing (see Table 1). The system is used to justify biowaivers – or formal exemptions from clinical BE and/or BA studies for a given drug product – for drug substances that demonstrate certain aqueous solubility and intestinal permeability characteristics. BCS was originally used to grant biowaivers for scale-up and post-approval changes for drug products, but was later extended to the approval of new generic products.
United States
The United States pioneered efforts to establish BCS as a regulatory tool in the drug approval process. In 2000, the US Food and Drug Administration (FDA) set forth a guide for sponsors to justify requests to waive in vivo BA and/or BE study requirements for IR solid oral dosage forms based on BCS criteria. The guidance provides recommended approaches for classifying an IR oral drug product and determining its dissolution characteristics. According to FDA, demonstration of BA or BE may not be necessary for substances that are highly soluble and highly permeable (i.e., fall into Class I) and exhibit rapid dissolution. More recently, the BCS has been used as an exemplar of FDA’s Critical Path Initiative efforts to employ more science-driven approaches to streamlining clinical trials.
However, some scientists have expressed concern that FDA has been too conservative in establishing dissolution, solubility, and permeability limits. Experts have proposed extending biowaiver eligibility to Class II drugs that are poorly soluble weak acids and to Class III drugs that exhibit rapid dissolution. Others have suggested that dissolution requirements be made less restrictive.
Europe
Introduced in 2001, the EMA guidance on the technical standards for biowaiver submission is slightly less stringent than that of the FDA. Under the latest version of EMA guidance (issued in 2010), IR oral dosage forms that demonstrate rapid dissolution and are highly soluble (i.e. fall into Class I or III) may be eligible to waive the in vivo BE requirement. In considering biowaiver applications, EMA places more importance on solubility than permeability; in vitro permeability data is accepted only in support of clinical data. To qualify for biowaiver application under the EMA guideline, drugs must be considered noncritical in terms of therapeutic range. Additionally, Class I drugs must not include excipients that are suspected of having any relevant impact on BA and Class III drugs must use excipients that are qualitatively the same and quantitatively very similar to those of reference products.
World Health Organization
Based on the FDA and EMA guidelines, the WHO set forth a guidance document for regulatory agencies around the world on the use of BCS-based biowaiver applications in 2006. The guide provides criteria for waiving in vivo studies for drugs featured in the WHO’s Model List of Essential Medicines. Subsequent analyses have shown that 63% of the WHO listed drugs fall into either BCS Class I or III (thus requiring only in vitro dissolution testing to establish BE). Since many top-selling drugs are not on the WHO Model List, a BCS-based provisional classification was applied to the top-selling 200 drugs of United States, Great Britain, Spain, and Japan and approximately 30% could be considered Class I. Under WHO’s guidance document, BCS-based biowaivers apply to Class I and Class III drugs products; more specifically, highly soluble active pharmaceutical ingredients with known human absorption/permeability characteristics are eligible for the BCS-based biowaiver approach for establishing the safety and efficacy of generic products. In addition, the WHO document also recognises the potential for biowaivers of Class II drugs that are weak acids. The WHO is less conservative than the FDA documents in terms of definition of high permeability of a drug, but a bit more stringent regarding solubility requirements.
What is the status of Biopharmaceutics Classification System (and BCS analogues) in Asia-Pacific?
To keep pace with global trends, Asia-Pacific countries have begun to introduce standards for waiving requirements for costly bioequivalence studies. While some of these countries currently maintain their own distinct systems for determining which drugs qualify for BE exemption, others have developed biowaiver guidelines based on BCS criteria or have proposed to incorporate BCS into their policies. In one way or another, each of the Asian regulatory agencies discussed below takes criteria derived from the concepts underlying BCS into consideration for biowaiver applications.
India
In 2005, India’s Central Drugs Standard Control Organization (CDSCO) issued Guidelines for Bioavailability and Bioequivalence Studies. Although these guidelines do not explicitly utilise BCS, CDSCO’s consideration of biowaiver eligibility is largely based on dissolution, solubility, and permeability criteria and closely resembles the technical standards set by the US FDA (see Figure 1). According to CDSCO, in vitro testing may replace in vivo testing when dissolution is very rapid and permeability and solubility are high; under BCS, this would refer to Class I IR solid oral dosage forms. Indeed, CDSCO has recently considered adopting BCS standards for determining biowaiver eligibility; they have proposed providing biowaivers to rapidly dissolving, highly soluble, highly permeable Class I drugs. The proposal to adopt BCS criteria would not appear to significantly alter the biowaiver system already in place in India.
Japan
 Like India, Japan maintains its own set of guidelines to justify BA/BE biowaivers. Japan’s National Institute of Health Sciences (NIHS) has established BA and BE biowaiver requirements for generic products, post-approval formulation, and dosing changes of existing drug products. NIHS establishes BE with the use of multimedia dissolution tests. Under this system, NIHS recommends that solubility must not be low, but does not consider permeability and does not set strict dissolution requirements for biowaiver eligibility. Biowaivers are not accepted under Japanese regulations for the first approval of generic drug products; for this, NIHS always requires in vivo bioequivalence testing. Overall, NIHS’s biowaiver requirements are less conservative than those of FDA, EMA, and WHO; the requirements allow for all BCS-based classes of drugs products to be considered for biowaiver and for in vitro qualification and allow for a larger range of formulation changes.
Like India, Japan maintains its own set of guidelines to justify BA/BE biowaivers. Japan’s National Institute of Health Sciences (NIHS) has established BA and BE biowaiver requirements for generic products, post-approval formulation, and dosing changes of existing drug products. NIHS establishes BE with the use of multimedia dissolution tests. Under this system, NIHS recommends that solubility must not be low, but does not consider permeability and does not set strict dissolution requirements for biowaiver eligibility. Biowaivers are not accepted under Japanese regulations for the first approval of generic drug products; for this, NIHS always requires in vivo bioequivalence testing. Overall, NIHS’s biowaiver requirements are less conservative than those of FDA, EMA, and WHO; the requirements allow for all BCS-based classes of drugs products to be considered for biowaiver and for in vitro qualification and allow for a larger range of formulation changes.
It is unlikely the BCS will ever be completely adopted to justify biowaivers in Japan; BCS has been viewed skeptically for regulatory purposes by Japan’s NIHS. NIHS maintains that formulation and manufacturing, rather than solubility and permeability, are indicative of bioequivalence. NIHS also states that while BCS serves to increase the use of dissolution tests in the US, BCS will actually decrease the use of these tests in Japan because they are already extensively applied. NIHS argues that permeability is still not known for many drugs, making it difficult to use BCS to establish regulatory bioequivalence for such drugs.
Russia
In order to establish the safety and efficacy of generic drug products in Russia, in vivo bioequivalence studies are typically carried out. In 2010, the Russian Federal Service on Surveillance in Healthcare and Social Development proposed that BE for generic products may be evaluated using in vitro testing in a draft guidance on BCS-based biowaivers. The Russian biowaiver procedure was developed according to the EMA, FDA, WHO, and the Health Department of Ukraine guidance documents. It sets forth specific biowaiver criteria for generic drugs by class, taking into consideration solubility, permeability, and dissolution characteristics along with the excipients used in the formulation and the possible risks associated with therapeutic index and adverse events.
China
With a large consumer base for drug products, and an estimated $8 million per year already being spent on bioequivalence studies by its pharmaceutical industry, China stands to benefit a great deal from use of biowaivers, especially as its drug industry expands in size and geographic scope. It was not until 2011, however, that a BCS-based biowaiver policy was put forth to evaluate IR solid oral dosage forms in China. Authors Ning and Qu-neng proposed that China adopt WHO’s in vitro dissolution criteria and suggest that Class I and Class III drug substances be eligible for biowaivers. The authors further suggest that excipients should be qualitatively the same and quantitatively very similar between the test and reference products. Offering an added caution, Ning and Qu-neng advise that the biowaiver approach should only be used when the potential benefits outweigh the risk of an incorrect biowaiver decision in terms of public health and individual patient risks. As of 2011, China’s State Food and Drug Administration regulations allow for bioequivalence exemption when the IR solid oral dosages forms under question are either generic drugs without excipients that may impact on drug absorption or Class I drugs that are undergoing post-approval changes.
South Korea
South Korea maintains a thorough system of determining biowaiver eligibility. Under the Guidance Document for BE Study, published in 2008, the Korean Food and Drug Administration (KFDA) allows comparative dissolution tests to replace BE studies for solid oral dosage forms. For example, a dissolution test can replace a traditional BE study when a solid oral preparation of a new strength has the same dosage form and API as an approved drug product. Dissolution tests may also replace BE studies when changes are made in the level of an excipient. The KFDA guidance document also establishes criteria for BE biowaivers based on solubility, permeability, and dissolution characteristics outlined by BCS.
Association of Southeast Asian Nations
In an attempt to establish a standardised basis for when BA and BE studies are necessary in drug registration among member states, the Association of Southeast Asian Nations (ASEAN) Consultative Committee for Standards and Quality (ACCSQ) issued guidelines on the Conduct of Bioavailability and Bioequivalence Studies in 2004. ACCSQ primarily considers the BCS criteria, along with non-critical therapeutic range, when evaluating applications for IR solid oral dosage forms. The guidelines also provide requirements for the design, conduct, and evaluation of BA and BE studies. ACCSQ suggests that member states – which include the IMS Tier 3 emerging markets of Thailand, Vietnam, and Indonesia – follow the ACCSQ document in conjunction with EU and ICH guidelines and regulations on good clinical practice.
Global Harmonisation
BCS is a simple, inexpensive, and reliable tool routinely used to establish regulatory guidelines on which to base BA and BE waiver applications. However, this system is not currently implemented by all regulatory authorities worldwide, nor is it uniformly applied among those nations that have chosen to base biowaiver guidelines on BCS criteria. It is believed that the importance of BCS as a regulatory tool will continue to increase over time. However, the cost and time-saving benefits of BCS-based biowaivers cannot be fully realised until differences among the regulatory bodies of the world’s major drug producing nations are reconciled. Lack of central databases, language barriers, and trademark certificates have also been identified as challenges to global harmonisation.
BOX001
BCS in Industry
Regulatory authorities are not the only ones implementing BCS; pharmaceutical manufacturers also make use of BCS throughout drug discovery and development processes. BCS helps sponsors determine what actions are needed to demonstrate the bioequivalence of a new formulation.
Companies can potentially save hundreds of thousands of dollars in costs, and several months of time in development, if bioequivalence studies are avoided. It has been estimated that the application of BCS can result in annual savings of $35 million for the pharmaceutical industry [4].
The practice of submitting BCS-based biowaivers has become more routine as industry has realised the benefits of the system. However, industry is not yet taking full advantage of BCS for a wide variety of reasons. Sponsors are sometimes reluctant to apply for biowaivers due to the perceived lack of certainty of acceptance by the regulatory agencies. Moreover, industry implementation of BCS may also be limited due to unnecessary barriers in existing guidelines, compartmentalisation of company resources, or a general lack of knowledge about BCS or the biowaiver process.
BOX002
Expert Interviews on BCS
The utility of the BCS is being recognised throughout the entire cycle of drug development according to a recent survey of 20 experts from generic and brand-name companies, government and academia conducted by the Tufts Center for the Study of Drug Development (Tufts CSDD), sponsored in part by a grant from Absorption Systems, LP.
The Tufts CSDD interviews suggest the need for global harmonisation. Interviews with key experts in industry, government, and academia suggest that in order to maximise its utility it will be necessary for the regulatory authorities of major drug-producing countries to harmonise in vitro bioequivalence requirements and provide a central repository of BCS determinations for global access (article in press).






