
Biosimilars are promising remarkable cost-savings for booming healthcare budgets. Yet the full potential can only be harvested when a number of significant markets, regulatory and clinical hurdles have been overcome globally.
The European Union (EU) and its medicines agency, the EMEA, have successfully addressed the regulatory challenges needed to fulfil the promise of biosimilars in Europe. Three years ago, in April 2006, the first breakthrough biosimilar marketing authorisation was issued to the human growth hormone Omnitrope® followed by 12 additional approvals to date.
Biosimilars as cost-savers and improved versions of originator, biopharmaceutical products could provide significant benefits for all involved in the process of providing better healthcare.
Translating the pharmacoeconomic potential of biosimilars into real improvements in access to expensive life-saving medicinal care remains a remarkable challenge that must be urgently addressed by healthcare authorities and policy makers.
Biosimilars today
Generic medicines have been successful in making healthcare more affordable and accessible. Ability to enforce generic substitution by many governments worldwide has helped contain the costs of prescription drugs. On a global level, generics are accounted for reducing the health costs for over 20 years, saving up-to US$100 billion annually.
Biopharmaceutical medicines account for around 10-15 per cent of the current pharmaceutical market, and the sector is outperforming the market as a whole in terms of growth.
In the US alone, the most lucrative biopharmaceuticals market by far, protein drug therapies today account for about 16 per cent of spending on prescription drugs. Furthermore, the biologics are growing at a much faster rate than their small-molecule counterparts. The lack of availability of lower-cost competitors (i.e. biosimilars, follow-ons) in the US is one of the factors contributing to the rapid growth in spending for these drugs.
Many of the patents for biopharmaceutical products developed in the last three decades of the twentieth century have already expired, thus offering the opportunity for the follow-on versions to be developed and introduced to the market, increasing the treatment choices for patients, physicians and payers.
Scientific developments and achievements in the field of genomics, proteomics and biomarker research are paving the path for new biologic therapies, promising to be significantly more effective compared to current treatment choices. The most recent findings suggest that approximately 30-40 per cent of drugs in the near future (2009-2012) are likely to be speciality drugs, their most significant component being biopharmaceuticals. It is becoming more and more obvious that the future of the pharmaceutical industry lies with biotechnology hence the expenditures for biopharmaceuticals are predicted to continue to expand by 15-20 per cent annually.
In the US, healthcare costs have reached an alarming level that makes most people see the need for change. At the annual BIO conference just recently held in Atlanta, the US healthcare reforms were addressed by the industry leaders. Among other issues, they were particularly concerned about biosimilar legislation that is heating the current political debate in the Congress.
However, the discussion so far has produced many more questions than answers about safety, clinical effectiveness and clinical data package required for biosimilars or follow-ons, the term most used in the US. Despite the complexity of surrounding scientific questions and issues, data exclusivity period gains most attention of the industry lobbying today.
However, the EMEA, our pioneer in developing the legal as well as the regulatory framework for biosimilars, has already successfully addressed and answered these questions. To date, the European Commission has granted market authorisation for as many as thirteen biosimilar products from various companies and under a wide range of different brand names. Biosimilar products have been successfully put into clinical practice across European Union Member States in the last three years.
Since the EU regulatory pathway was put in place in 2005, just three biological molecules have been approved, growth hormone, erythropoietin and colony stimulating factor namely. An impressive number of thirteen biosimilar marketing authorisations granted so far is thus based on six biosimilar development programmes for just three different substances, see Table 1.
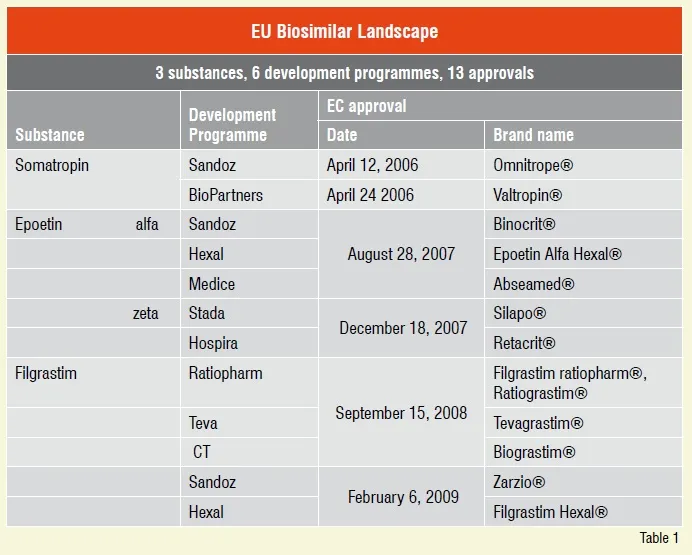
This did not happen overnight. European debate leading to the present regulatory approach took more than five years of discussions and consolidations of all stakeholders interested in biosimilars – the EMEA and its Working Parties, biological science, medical science, legal and regulatory experts, as well as pharmaceutical industry.
Pitfalls in European biosimilar developments so far? yes...
Three biosimilar development programmes have been either rejected by the EMEA or applications withdrawn from further evaluation process by the respective applicants. Developments of a biosimilar interferon alfa-2a, interferon beta-1a and human insulin have not yet delivered the expected result, an approval of a biosimilar product to compete in this ever more lucrative market.
Biosimilar legislation is evolving rapidly in other countries as well. Other national authorities have already successfully addressed the biosimilar regulatory and legal framework, among them Australia, Canada, Malaysia and Japan.
However, progress in the US, the most lucrative biopharmacueticals market with two thirds of global biological sales, is still painfully slow.
The science is proven and interesting
Biopharmaceuticals are medicines whose active substances are made by or derived from a living organism therefore these medicines are produced using biotechnology. They are proteins (including antibodies), nucleic acids (DNA, RNA or antisense oligonucleotides) used for therapeutic or in vivo diagnostic purposes, and are produced by means other than direct extraction from a native (non-engineered) biological source.
Biological medicinal products with patents already expired are all proteins of recombinant DNA origin. Proteins are generally large, complex molecules that mostly show heterogeniety which is a result of so-called posttranslational changes. Moreover, the biological activity of proteins usually depends on the 3-dimensional structure that must be maintained throughout manufacturing process, fill-finish as well as storage over the shelf-life of the final product dispensed to the patient.
Generic medicines must prove bioequivalence with the reference product in addition to showing chemical identity of the active substance to be considered therapeutically equivalent.
Authorised biosimilars have been compared to and have demonstrated that they match the reference product in terms of quality, efficacy and safety albeit they are not automatically considered as therapeutically equivalent.
Why is this?
All biopharmaceuticals are inherently variable due to the fact that they are produced from a living organism. Variability therefore exists within batches, batch-to-batch, after changes have been implemented to the previous production process, as well as between different manufacturers.
Current analytical techniques coupled with ‘quality by design’ approach in development and manufacture of a biosimilar medicine allow for a thorough comparability exercise and very close match of the quality parameters of the biosimilar compared to the reference. Yet all potentially clinically relevant differences between the biosimilar and the reference may not be detected during in-vitro physicochemical and biological comparability, therefore additional PK/PD studies as well as pivotal clinical trials in patients providing data on similar efficacy and safety profiles, are required in Europe.
For all biopharmaceuticals, originator and biosimilars, additional post-approval requirements such as pharmacovigillance as well as presentation of post-marketing safety data collected in respective surveillance studies, are mandated by the EMEA.
Market of the future
Most generic manufacturers are actively involved, either directly or indirectly, in the development of biosimilars, the alternatives of the original brand which should provide remarkable cost savings and better accessibility to state-of-the-art biopharmaceutical medicines.
But, will physicians use them? Ideally for generic manufacturers they will regard them as therapeutically equivalent and therefore substitutable by pharmacists, in countries where this is an option. The overarching question for physicians is going to be safety and efficacy; they need to be confident about a new alternative’s capabilities in order to switch.
For developers of biosimilars, the issue of interchangeability becomes a key point. In Europe, the biosimilar pathway leading to biosimilar approvals, speaks of ‘therapeutic similarity’ and leaves it at that. There is neither a claim on therapeutic equivalence nor on interchangeability. Therapeutic equivalence is going to be a hard nut to crack in regulated markets. Many of the biosimilar bills before congress in the US already encompass specific provisions which allow applicants to try to demonstrate this, but a clear view from the FDA is currently not yet available.
Another challenge ahead of us is substitutability i.e. can the pharmacist provide a biosimilar version if the physician has prescribed the original brand?
What could be the future role of the insurers, payers, could they mandate substitution to manage their available budgets?
Currently, access for patients to biosimilar medicines in the EU is not automatic, it still requires proactive steps to be taken by all relevant stakeholders, including calling on the physicians to obtain prescription. Issues of reimbursement are left to each individual Member State. There is little public sign that governments have begun to address the market pathway for biosimilars to enable access to these medicines as soon as possible after their marketing approval. Quite the contrary, countries such as France, Italy, the Netherlands and Spain, for example, have explicitly stated that biosimilars will not be substitutable, while the UK government has indicated that it does not expect substitution to occur as brand prescription is mandated for all biopharmaceuticals including biosimilars.
Conclusions
Research and development investment in new medicines by the biopharmaceutical industry stood at US$ 65.2 billion in 2008. Branded biologics have, until recently, been immune to generics competition. However, the breakthrough approval of biosimilars in Europe marks a changing landscape in the realm of biopharma. Through the collective effort of all stakeholders important scientific and legal questions of biosimilars have been successfully addressed in the EU.
Biopharmaceuticals still remain some of the world’s most expensive medicines. Many of them providing significantly improved outcomes in difficult-to-treat diseases such as cancer and autoimmune disorders, or successfully replacing and supplementing a protein normally produced by the healthy body.
Biosimilars are, therefore, vital to ensuring the sustainability of healthcare systems. Where available today, they increase choice in access to state-of-the-art biopharmaceuticals, providing benefits to all stakeholders including patients, physicians and healthcare providers.
References
1. 2009 Medco Drug Trend Report. (Accessed June 2, 2009 at: http://medco.mediaroom.com/index.php?s=64&cat=5)
2. EMEA. EPARs for authorised medicinal products for human use. (Accessed June 1, 2009 at: http://www.emea.europe.eu/htms/human/epar/a.htm)
3. Rader RA, (Re)defining biopharmaceutical, Nat Biotechnol 2008; 26: 743-51. (Accessed June 2, 2009 at: http://www.biopharma.com/biopharmacopeia//redefining.pdf)





