
The SARS-CoV-2 pandemic has required new methods for rapidly developing effective vaccines and drugs to treat COVID-19 patients. There are approximately 12,000 existing drugs, approved natural products, and clinical trials candidates whose safety in man has already been established. Computational repurposing of these drugs, where mathematical methods are used to simulate their interactions with key proteins in the virus, is a rapid way to find new treatments for SARS-CoV-2.
Introduction
The COVID-19 pandemic caused by the SARS-CoV-2 virus has infected at least 500 million people and has killed almost 7 million, both numbers likely to be underestimates. There has been a massive mobilisation of scientific research efforts around the world to understand the origin and structure of the virus, and to develop effective vaccines and drugs to treat patients with COVID-19, the illness caused by the virus. While rapid progress has been made in a relatively short space of time in both vaccines and therapeutics, the treatment options for patients severely ill with COVID-19 remain limited. Unfortunately, traditional development of new drugs takes of the order of 10-15 years, which is unacceptably long for pandemics when drugs are needed as soon as possible.
One approach to drug discovery that can be productive in very short time frames is the repurposing of existing drugs. Already approved drugs, natural products, and drugs already in clinical trials for other indications have all been tested for safety in man and could be used ‘off label’ for emergency use in a pandemic. The principle behind our in silico antiviral drug screening approach is that all drugs hit targets other than those for which they are designed, this off-target activity is the origin of most drug side-effects. This off-target activity has also been exploited in the serendipitous discovery of several blockbuster drugs such as Viagra and Minoxidil (both originally antihypertensive drug candidates). While all 12,000 or so repurposing candidates could in principle be tested experimentally for activity against virus targets or in vitro, it is much faster and cheaper to use computational methods to achieve this. This realisation caused an unprecedented surge in publications where we and others have used molecular docking to identify promising drugs that could bind to the dozen or more viable target proteins in SARS-CoV-2.
While most widely used molecular docking software is easy to run, these studies have varied enormously in quality and outcomes because, without full understanding of the protein target properties and limitations of the software, the results obtained can be unreliable at best and meaningless at worst. Considerable understanding of the structural biology of the targets and the operational parameters of the docking software is required to achieve robust results, as was reviewed very recently.
In the studies summarised below, rigorous computational methods were used to identify potentially useful COVID-19 drug candidates for repurposing. This involved a combination of validated molecular docking methods (that calculate the orientation and binding energy of drugs to binding sites in proteins), and molecular dynamics methods (that allow for protein and drug flexibility and calculate more accurate binding poses and energies) to identify the most promising drugs and natural products for treating COVID-19. Most importantly, rigorous quality control measures were applied at every step in the process to ensure the quality of the target predictions.
Three key protein targets essential for the function of SARS-CoV-2 were used to screen a large library of ~12,000 small molecule drug candidates. The targets were the main protease of the virus, Mpro or 3CLpro, the RNA -dependent RNA polymerase (RdRp, essential for viral replication), and the helicase (also part of the virus’ replicative machinery). The docking calculations were validated by redocking drugs into the relevant protein binding sites to recapitulate the binding poses in the experimental structure. The calculations were run on large GPU cluster machines generously made available during the pandemic by Oracle Cloud Systems. The top 80-100 drug candidates identified by the docking calculations were subjected to molecular dynamics calculations to improve the estimates of the relative binding energies of drugs to proteins, and their binding poses in the protein active sites. Subsequent literature searches disclosed that >30 per cent of our predicted repurposing candidates for all three SARS-CoV-2 protein targets have experimental validation data, a very impressive validation rate for the computational methods employed.
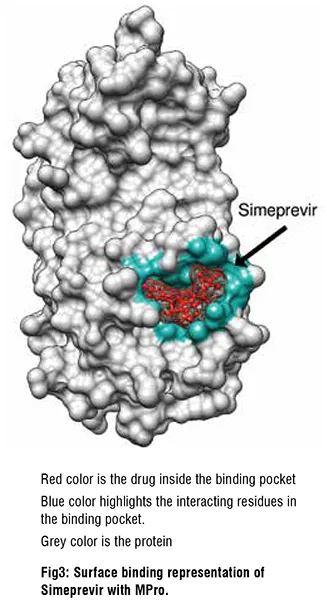
SARS-CoV-2 Mpro inhibitors
Mpro is a key protease essential to SARS-CoV-2 function. We therefore screened drug libraries for compounds that might bind and inhibit Mpro . Several of the top ten best binding drug candidates were antiviral agents simeprevir, sofosbuvir, lopinavir, ritonavir and remdesivir. The more interesting candidates had similar binding affinities and were of diverse activity classes. These were bemcentinib (AXL receptor kinase inhibitor), montelukast (leukotriene receptor antagonist), ergotamine and mergocriptine (ergot alkaloids with adrenergic and dopaminergic receptor agonist activities). Bemcentinib shows in vitro activity against SARS-CoV-2 with 10-40 per cent protection at 50μM in Vero cells and an IC50 of 100nM and CC50 of 4.7μM in Huh cells. Indeed, it is already an investigational treatment for COVID-19 , with a phase 2 trial underway. Montelukast another interesting hit has been shown to produce a significant reduction in SARS-CoV-2 infection in elderly asthmatic patients and has a reported in vitro SARS-CoV-2 IC50 of 18.8 μM, and CC50 >20 μM.
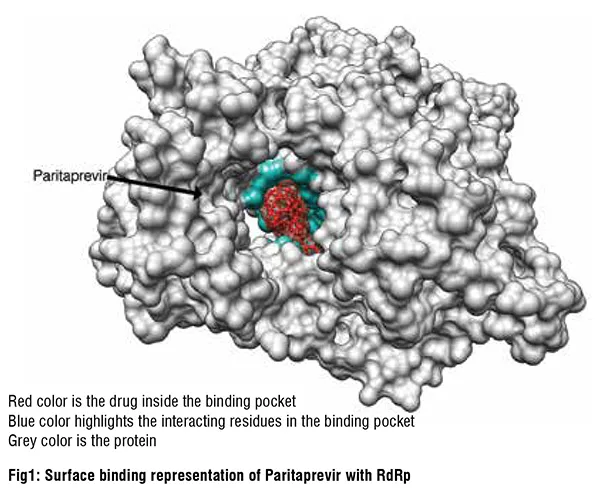
SARS-CoV-2 RdRp inhibitors
Given the function of SARS-CoV-2 RdRp to bind RNA, its binding site is large. Most of the repurposed drugs in the list of 20 best binders we predicted had relatively large complex structures and substantial ligand flexibility. Antiviral drugs accounted for half of this list, the remaining compounds in being mostly natural products or their derivatives. These were of particular interest given their relative novelty and molecular diversity. Interestingly, our computational screen identified ivermectin as fitting snugly into the RdRp binding pocket. Ivermectin and other avermectins and milbemycins are broad-spectrum antiparasitic macrocyclic lactones that have shown inconsistent activity against SARS-CoV-2. Ivermectin has been widely touted as a drug against COVID-19, although the debate around its use has become highly politicized. It is interesting, therefore, that our unbiased screening methods identified Ivermectin as one of its top hits. Digoxin is a widely used cardiac drug used to treat heart arrythmias and cardiac failure and has a large complex structure that fitted well into the RdRp binding pocket. It exhibited potent in vitro antiviral effects against SARS-CoV-2 in Vero cells (IC50 37 nM). Another tightly binding natural product, silibinin, is a flavonolignan that is the major active constituent of silymarin, a standardised extract of the milk thistle seeds. It can function as an antioxidant, antineoplastic drug, and hepatoprotectant. Rapamycin, a macrolide antifungal metabolite with immunosuppressant activity that also bound tightly to RdRp, is currently in COVID-19 clinical trials. Other highly ranked natural products from our in silico screens included: carbetocin, a synthetic analogue of oxytocin;eribulin, a fully synthetic macrocyclic ketone analogue of the marine natural product halichondrin B, a potent anti-mitotic anticancer agent; novobiocin, a DNA gyrase and bacterial type IIA topoisomerase inhibitor; and ergotamine and related ergot alkaloids also predicted by us and others to bind several SARS-CoV-2 molecular targets, including the main protease, Mpro.
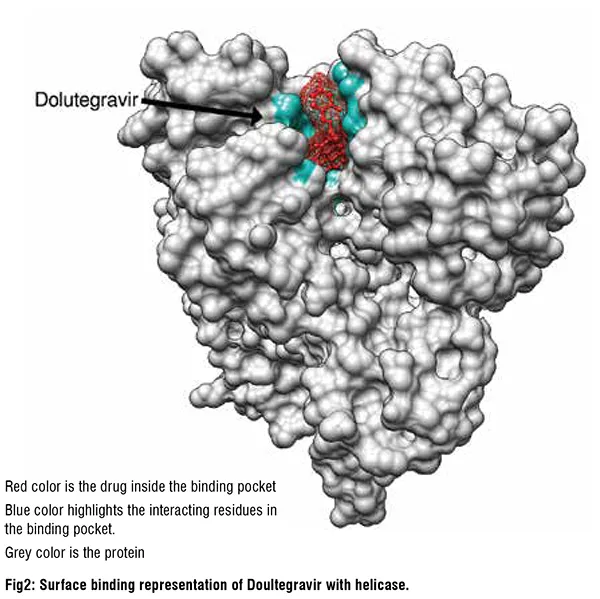
SARS-CoV-2 Helicase inhibitors
The twenty molecules with the best predicted helicase binding affinity can be broadly characterized as containing one or more hydrophobic aromatic moieties linked to another polycyclic moiety containing hydrogen bond donors or acceptors. These molecules come from diverse drug classes, with antiviral agents making up 25 per cent of the hits and antihistamines and antipsychotics also being well represented. The most interesting non-antiviral agents identified by the study were hesperidin(citrus flavanone glycoside), rutin (flavonol glycoside), conivaptan (vasopressin inhibitor), aprepitant (NK1 antagonist), manidipine (Ca channel blocker, anti-hypertensive), aminoquinuride (trypanocidal agent), antrafenine (analgesic anti-inflammatory), epirubicin (anticancer intercalator), dicoumarol (anticoagulant), fluspirilene (antipsychotic), fexofenadine (antihistamine), astemizole (antihistamine), sertindole and pimozide (antipsychotics). Of these manidipine exhibits broad-spectrum antiviral activity, with SARS-CoV-2 IC50=2μM in HUH7 cells and 7.5μM in Vero cells. Tipranavir also exhibits a relatively broad spectrum of antiviral activity, inhibiting replication of SARS-CoV-2 in VeroE6 cells. Fluspirilene activity against SARSCoV- 2 was reported to be 3.1μM in Vero E6 cells, mirroring the activity of fluspirilene against MERS-CoV and SARS-CoV in Vero E6 cells of 7.5μM and 6.0μM, respectively. Astemizole had an EC50 of 1μM against SARSCoV- 2 in Vero E6 cells and 4.9μM and 5.5μM against MERS-CoV and SARS respectively.
Conclusions
Computational screening methods promise fast and reliable identification from libraries of existing drugs, clinical trials candidates, and approved natural products of compounds that may have useful activity against SARS-CoV-2. The three studies summarised here show that docking methods are capable of reliably predicting the binding pose of drugs to structures of virus target proteins. ComplementaryMD simulations performed on the repurposing candidates with the highest docking scores provide more reliable relative binding affinities of these drugs for the targets, allowing the docking hits to be ranked for activity. This is a very rapid and efficient way to screen for potential anti-viral drug activities.
The fact that >30 per cent of the top 80-100 repurposing candidates we identified have experimental activity against the target and/or the virus, with several already in clinical trials for treating COVID-19 shows the computational predictions were highly reliable. Therefore, the 70 per cent of repurposing candidates identified by these studies that have not yet been assessed for in vitro or in vivo activity could provide a rich resource of potentially useful drugs for treating COVID-19. As these are generally applicable, platform methods, they are equally suitable for identifying repurposed drugs to treat neglected tropical diseases, or viral infections and future pandemics.[5]We are seeing the beginning of an era where experimental high throughput drug screens will complement structural biology-based and machine learning methods to provide faster, more efficient, cheaper, and more robust methods for repurposing existing drugs or discovering new ones.
References
1. Muratov, E.; Brown, N.; Fourches, D.; Kozakov, D.; Medina-Franco, J.L.; Merz, K.; Isayev, O.; Oprea, T.; Poroikov, V.; Varnek, A.; Winkler, D.A.; Zakharov, A.; Cherkasov, A.; Tropsha, A. A critical overview of computational approaches employed for COVID-19 drug discovery. Chem. Soc. Rev., 2021, 50, 9121-9151.
2. Piplani, S.; Singh, P.; Petrovsky, N.; Winkler, D.A. Computational Repurposing of Drugs and Natural Products Against SARS-CoV-2 Main Protease (M pro ) as Potential COVID-19 Therapies, Front. Mol. Biosci. 2022, 9, 781039.
3. Piplani, S.; Singh, P.; Petrovsky, N.; Winkler, D.A. Computational screening of repurposed drugs and natural products against SARS-CoV-2 RNA-dependent RNA polymerase as potential COVID-19 therapies, Mol. Biomed. 2021, 2, 28
4. Piplani, S.; Singh, P.; Petrovsky, N.; Winkler, D.A. Potential COVID-19 therapies from computational repurposing of drugs and natural products against the SARS-CoV-2 helicase, RSC Digit. Dis. 2022 in review.
5. Winkler, D.A. Use of artificial intelligence and machine learning for discovery of drugs for neglected tropical diseases, invited paper, Front. Chem. 2021, 9, 39







