
COMPUTER AIDED DRUG DESIGN IN PHARMA R&D
Mallikarjuna Rao Pichika, International Medical University
Mak Kit-Kay, International Medical University
The number of new US FDA approved drugs is declining in spite of lots advances made in science. This fact signifies the complexity of drug development process. Pharma companies are closing down their R&D efforts and hoping that recent advances in computational techniques provide a solution to improve the efficiency.
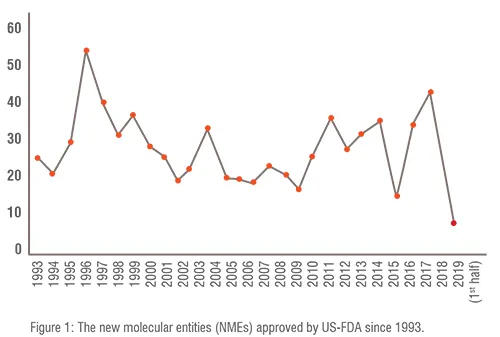
Pharma R&D landscape : The number of New Molecular Entities (NMEs) approved by the United States Food and Drug Administration (US-FDA) is shown in Figure 1. NME is defined as medication containing an active ingredient that has not been previously approved for marketing in any form in the United States. Although the number of US-FDA approved NMEs per annum has remained constant over the years, pharma R&D efficiency (NMEs approved per billion US$ spent on R&D) is declining. The reasons for this decline could be attributed to stringent requirements imposed by US-FDA especially after disastrous thalidomide incident. During the 1960s thalidomide was marketed as a mild sleeping pill. However it caused thousands of children to be born with malformed limbs.
Drug Discovery Strategies
The main chronological strategies that have been used in drug discovery are phenotypic screening followed by target-based screening and drug repurposing. Phenotypic screening involves testing of a large number of randomly selected compounds in a systems-based assay. Target-based screening involves manipulation of a selected enzyme or receptor to produce a desired therapeutic response. In general, the minimum duration of drug discovery cycle from concept to market in either phenotypic or target-based screening is 10 years while it takes only three years in drug repurposing.
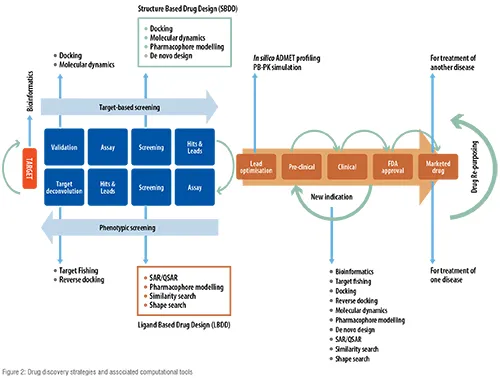
Recently, Big Pharma have tended to incline towards computational techniques for the discovery of innovative drugs with the hope of improving the R&D efficiency. The two main approaches in computer aided drug discovery (CADD) are structure-based drug discovery (SBDD) and ligand-based drug discovery (LBDD), which could be applied in all the aforementioned drug discovery strategies. In SBDD, three-dimensional (3-D) structure of protein is analysed to identify potential binding sites and key interactions producing respective pharmacological activities. Using this information, attempts are made to discover novel drugs with high potency and selectivity. In LBDD, the structure of target protein is unknown and it focusses on chemistry of bioactive ligands to develop a structure-activity relationship between physiochemical properties and bioactivities. Using this information, novel ligands will be designed with improved bioactivity. Figure 2 shows the various computational tools used in all the three types of drug discovery strategies.
Structure-based Drug Discovery (SBDD)
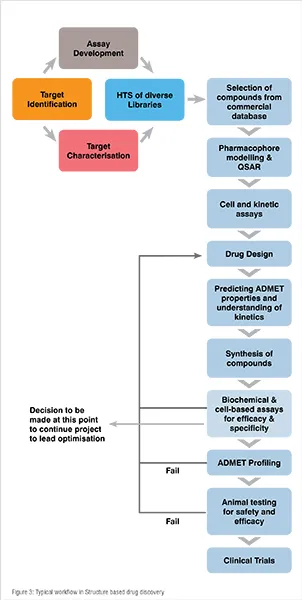
This approach is used if both the target protein structure and ligand are known in which the ligand inhibits the activity of the protein through competitive binding mechanism. It is neither a single tool nor technique but it a mixture of both experimental and computational techniques. It has the highest success rate, therefore it is a preferred method of CADD approach. A typical flow chart of activities in SBDD approach is shown in Figure 3.
Ligand-based Drug Discovery (LBDD)
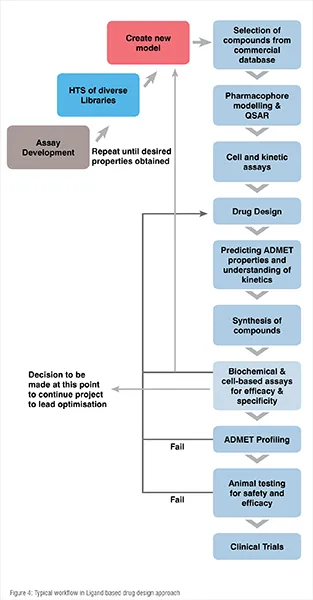
This approach is used if the target protein structure is not known but the ligand structure is known in which the ligand modulates the function of the protein. It has a lower success rate compared to that of SBDD. It is a mixture of both experimental and computational techniques. A typical flow chart of activities in LBDD approach is shown in Figure 4.
Representative Success Stories of CADD
A binding pocket for a new class of drugs to treat AIDS was discovered using docking while considering the flexibility of the receptor through molecular dynamics. This information leads to discovery of orally available HIV integrase inhibitor, raltegravir (Isentress®), approved by FDA in 2007 and received approval for paediatric use in 2011.
The same molecule (HTS-466284), a 27nM inhibitor, was discovered independently using virtual screening by Biogen IDEC and traditional enzyme and cell-based high-through put screening by Eli Lilly. The computational work involved pharmacophore-screening of 200,000 compounds and used as a starting point. The compound discovered experimentally at Lilly required in vitro screening of a large library of compounds to find potential inhibitors in a TGF–ß–dependent cell-based assay and chemical synthesis.
A CADD program (homology modelling, virtual screening, hit to lead optimisation and in silico profiling) led to clinical trials of a novel, potent, and selective anti-anxiety, anti-depression 5-HT1A agonist in less than 2 years from the start and requiring less than 6 months of lead optimisation and synthesis of only 31 compounds.
Applying QSAR algorithms to toxicity data and corresponding chemical structures led to the development of in silico tools that predict toxicity response (mutagenicity, carcinogenicity) and toxicity dosing (no observed effect level, NOEL; maximum recommended starting dose, MRSD).
Aggrastat (Tirofiban), from Merck, a GP IIb/IIIa antagonist (myocardial infarction, it is an anticoagulant and platelet aggregation inhibitor, protein-protein interaction inhibitor) results from a lead compound that was further optimised using ligand-based pharmacophore screening.

Zanamivir is a neuraminidase inhibitor (transition-state analogue inhibitor) used in the treatment and prophylaxis of influenza caused by influenza A virus and influenza B virus. Zanamivir was the first neuraminidase inhibitor commercially developed and It is currently marketed by GlaxoSmithKline under the trade name Relenza as a powder for oral inhalation.

Conclusion
CADD, combined with biophysical approaches and HTS, does help the drug discovery process. CADD approaches assist in decision making, contribute to improve the R&D efficiency, generate new ideas and concepts, suggest solutions to problems and rapidly test the hypothesis. They allow the investigation of the activity of new compounds before they are even synthesised. CADD approaches also help to analyse millions of heterogeneous data points originating from multiple sources. They assists in identifying the either molecular mechanism or polypharmacology or ADMET properties of a compound. Now-a-days, there is a massive improvement of data mining and artificial intelligence technologies for systematic analysis of massive data that is being generated by both experimental and computational experiments. Analysis of 142 drug discovery and development projects from Astra Zeneca revealed five critical factors (5R frame work; Right Target, Right Tissue, Right Safety, Right Patients and Right Commercial Potential) that could improve the R&D efficiency.
Author Bio

Mallikarjuna Rao Pichika, Professor, Associate Dean (Research & Consultancy) and Head of Centre of Excellence for Bioactive Molecules and Drug Delivery at Internatoinal Medical University, Kuala Lumpur, Malaysia. He is specialised in medicinal chemistry with special interest in computational drug discovery approaches. He authored numerous number of research publications and hold few patents.

Mak Kit-Kay, Lecturer in Pharmaceutical Chemistry department with lots of enthusisam in Artificial intelligence. She is the recipient of Wellcome Trust Fellowship from Wellcome Centre for Anti-infectives Research and Artificial intelligence molecular screen (AIMS) award from Atomwise. She is one of the top 3 contestants in the BioSolveIT Scientific Challenge.




