
Poorly soluble drugs are often formulated in lipid-based Self Emulsifying Drug Delivery Systems (SEDDS) to improve their oral bioavailability. These SEDDS are multi-component formulations containing lipids, surfactants, and polar organic solvents necessary to solubilise the drug. One key factor affecting the performance of these formulations is the solubility of the drug in the excipient mixture. Traditionally drug solubility in the excipient mixture is assessed by equilibrating excess drug in the mixture and then the final formulation is prepared at sub-saturation concentration of the drug. Interactions between the drug and lipid excipient in the formulation could lead to formation of molecular complexes that may be less soluble than the drug itself. In such cases, determination of drug solubility is complicated due to the presence of the complexing excipients and the solubility of the drug will depend on the equilibrium relationships among those species involved and the concentration of the species in the excipient mixture. Because of the drug-excipient interactions and consequently, the precipitation of the complex during a solubility experiment, the equilibrium drug concentration will vary depending on the relative amounts of drug and the complexing excipient initially added to the solubility samples. Although it is preferred to avoid using any complex-forming excipients, they may often have to be considered due to their unique functionality necessary for the performance of the SEDDS formulation.

Currently, information is not available with regard to understanding the solubility properties of poorly soluble drug-excipient complexes in lipid-based SEDDS. Such information would be important since many lipid-based formulations consist of multiple excipients, raising the chances that drug-excipient interactions resulting in complexation may occur. Understanding of the solubility behaviour of the drug-excipient complex in such a system will help formulation scientists in rational selection of the appropriate excipients and in the determination of the optimum excipient levels to ensure drug remains solublised.
The purpose of this report is to describe the solubility properties of a poorly soluble drug-excipient complex in a SEDDS formulation and to introduce an approach for evaluating the equilibrium drug concentration in such a system. Tipranavir (TPV) was used as a model compound. TPV is a novel non-peptidic protease inhibitor that is currently on the market for treating HIV positive patients. The compound is a weak acid (pKa 5.5), having very low aqueous solubility (0.1 ug/mL in pH 2 and 17 ug/mL in pH 7.5). It is highly lipophilic (log P = 6.0) and has high solubility in various polar organic solvents, surfactants and lipidic excipeints. TPV was found to form a poorly soluble complex with 1,3 dioctanoylglycerol (DOG), a component of Capmul MCM® which is a widely used lipid excipient. The TPV-DOG complex has a 4:1 TPV-to-DOG molar ratio.
Preparation, characterisation of TPV-DOG complex crystals and solubility determination
Sufficient amount of the TPV-DOG complex was prepared by dissolving TPV and DOG in ethanol at 60°C followed by evaporation of ethanol. The formation of the complex with a 4:1 TPV-to-DOG molar ratio has been confirmed by XRPD, DSC, and NMR. The complex is a crystalline material and has a different melting point from the TPV itself. Total TPV solubility was measured by equilibrating excess amount of TPV-DOG complex in the SEDDS vehicles containing various amount of DOG at 25°C. Samples were ultracentrifuged, filtered through a 0.45 um filter and then analyzed by HPLC for total TPV concentration. Characterization of the solid materials isolated at the end of the solubility study indicates that the complex did not change form during the solubility study.
The experimental data of total TVP solubility shown in Figure 1 are fitted to Eq. 7 by non-linear fitting using Micromath Scientist Version 3.0. Values of So and K41 are simultaneously estimated to be 0.0186 ±0.0025 (M) and 21.97±7.19 (1/M4), respectively. The fitted curves are in good agreement with the experimental data, as shown in Figure 1. This result indicates that the proposed equilibrium relationships in Scheme 1 are appropriate for describing the solubility properties of the complex.
Prediction of equilibrium TPV concentrations and precipitation of the complex in SEDDS formulations
With the known values of So and K41, the equilibrium concentration profiles of the total and free TPV, and free DOG as a function of the total DOG in the SEDDS solution can be calculated using Eqs. 5-7, respectively. The predicted concentration profiles are shown in Figure 2. These concentration profiles provide an insight in understanding the solubility properties of the complex and the relationships among the various complexing species in the system.
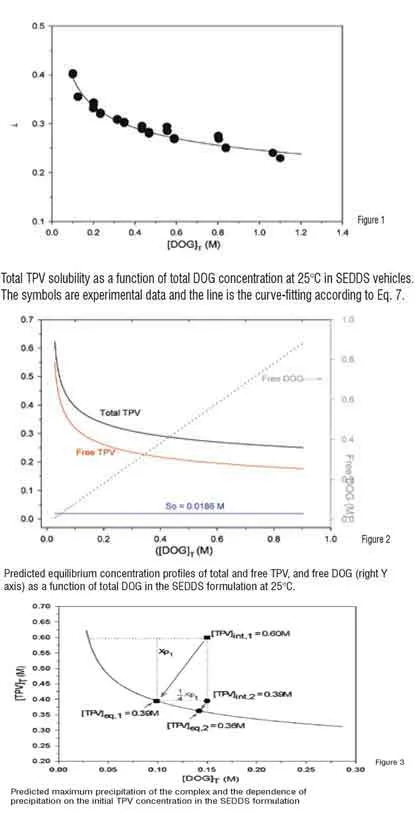
DOG is a component of Capmul MCM and the weight fraction of the DOG in Capmul can be determined by NMR. Therefore, Eq. 7 can be used to predict the equilibrium TPV concentration at any given level of Capmul MCM. This prediction provides a useful tool in selection of appropriate level of Capmul MCM in the SEDDS to avoid precipitation of the TPV-DOG complex.
In the case where the drug content is above its equilibrium concentration in the SEDDS containing a given amount of DOG, precipitation of TPV-DOG complex will occur. The amount of precipitation can be predicted based on Eq. 7. When precipitation occurs, both TPV and DOG will decrease from the solution in a 4 to 1 molar ratio. As a result, this concentration decrease should stop when a new equilibrium is established where the concentrations of TPV and DOG satisfy the solution complex constant (K41) and the solution complex concentration ([TPV4:DOG]soln) is So.
Assume the maximum loss of TPV as a result of TPV-DOG complex precipitation is Xp (molar concentration), we have,
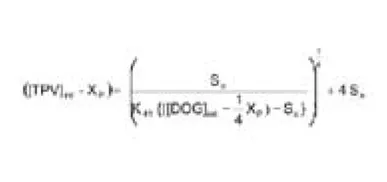
Where [TPV]int and [DOG]int are the initial molar concentrations of TPV and DOG in the SEDDS, respectively. Given the known values of So and K41 and the initial TPV and DOG concentrations, the value of Xp can be calculated using Eq. 8. As an example shown in Figure 3, in a SEDDS vehicle containing initial [DOG] = 0.15 M, the TPV concentration at [TPV]int,1 = 0.60 M is above the saturation line (supersaturated) and will precipitate out as the TPV-DOG complex. As a result, the TPV concentration will drop to reach the new equilibrium at [TPV]eq,1 = 0.39 M, with a maximum precipitation of Xp1 = 0.21 M. At this new equilibration, the DOG in the SEDDS is consumed by (1/4 Xp1) = 0.05 M.
The drug concentration -dependent solubility of TPV observed when the solubility was performed starting with the TPV powder in the Capmul containing SEDDS vehicle, as mentioned in earlier sections, can now be further explained using the examples in Figure 3. In a SEDDS vehicle containing 0.15 M of DOG, if the TPV powder added to the solubility sample is targeted at 0.60 M ([TPV]int,1), the measured final equilibrium concentration of TPV will be 0.39 M ([TPV]eq,1). However, if the initial TPV powder added is 0.39 M ([TPV]int,2) (still above the saturation line, but with a less excess amount of TPV powder), the final equilibrium concentration of TPV will be 0.36 M ([TPV]eq,2). This is because at the end of equilibration, the solid in equilibration with the solution will be converted from the initial TPV to the TPV-DOG complex. The higher initial amount of TPV, which depletes more DOG from the solution, results in a higher final equilibrium concentration of TPV.
In conclusion, the solubility properties of the TPV-DOG complex in the SEDDS can be described by two equilibria: a liquid-solid phase equilibrium of the complex and a species equilibrium in the liquid phase. The two parameters, So and K41, can be determined by curve-fitting the solubility data into the model equation. The model was in good agreement with experimental results. The model equation can successfully predict the concentrations of total and free TPV at any given level of DOG in the formulation. The approach developed provides a useful tool for rationale selection of excipients and their levels to avoid drug precipitation in lipid-based drug delivery systems.
Acknowledgements
Mayur S. Dudhedia, Zeren Wang, Richard T. Darrington, Toby Tamblyn, John A. Smoliga, Paul-James Jones, and Steven L. Krill are acknowledged for ontributions to this work. Fernanda Villagra and Francisco Gel are thanked for providing the reference complex material, Rosalie Ferrillo and Corrine Jankovsky for enerating the XRPD and DSC data, and Maimoona Chinwala for generating some of the HPLC data. Nair Rodriguez-Hornedo and Bradley D. Anderson are acknowledged for helpful discussions regarding this research project.





