
Biopharmaceuticals can be broadly defined as drug substances obtained from a living organism using biotechnology. Typically, biopharmaceuticals are large molecules with complex structural features and conformations, compared to small molecule drugs. In 2005, biologic drug sales were about US$ 32 billion comprising 14 per cent of total drug sales, and this number is expected to climb to 20 per cent by 2010. This translates to a 17 per cent growth year to year for biopharmaceuticals, compared to 5.7 per cent increase for small molecule drugs.
Projected sales of biotechnology derived products are expected to reach US$ 90 billion this year alone. There are currently about 125 biopharmaceuticals marketed worldwide and approximately 633 new products are in the pipeline, making up over 19 per cent of all new drug development programmes. Twenty per cent of currently marketed blockbuster drugs (sales > US$ 1 billion) are biotechnology-derived.
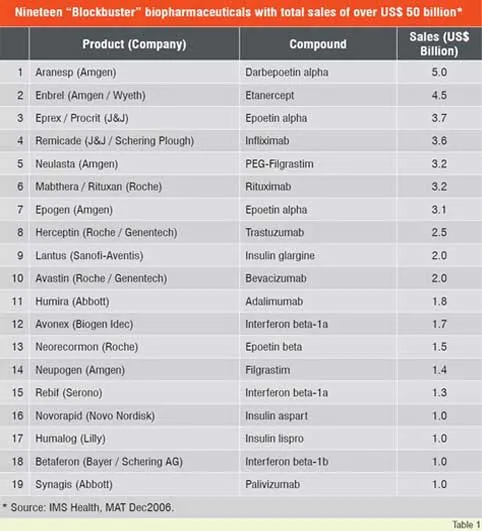
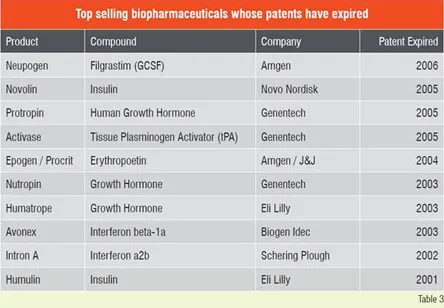
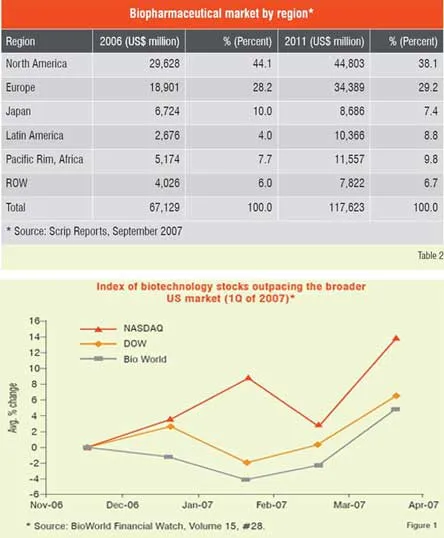
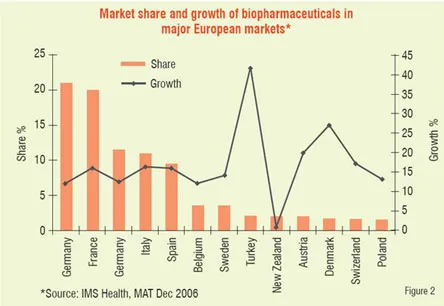
Number of biologic blockbuster medicines has increased over the years. There were 20 products of over US$ 1 billion sales in 2006 compared to only 3 products in 2000. The 19 top selling biologicals account for about US$ 50 billion of the total market (Table 1). Remarkable performance growth is expected in the coming years from biotechnology-based companies as seen from Figure 1, which depicts an index of biotech stocks outpacing the broader market indices in 2007. The same trend is shown in Figure 2 for Europe with market share of major EU countries and their growth potential. The current anticipated global markets for biopharmaceuticals is shown in Table 2, and as expected North America and Europe are expected to garner a combined market share of over 67 per cent.
This impressive growth in biologics has attracted top-tier generic industries to the biogenerics arena. Despite the significant costs and hurdles expected in developing and gaining approval for such products, the business prognosis for biogenerics remains attractive. The leading generic companies with significant biologic portfolios include Teva, Sandoz, Barr / Pliva, Stada, Mayne, Ratiopharm, Biocon, Cangene, BTG and Wockhardt. Attractive features for investing in biogenerics include higher profit margins, lower price erosion, longer market share retention and lower competition through high barrier to entry. On the other hand, development of biogenerics entails several risk factors, including high development costs and uncertain regulatory and legal landscape. Several of the top selling biopharmaceuticals (Table 2) have already lost their patent protection, and several more are expected to lose their exclusivity in the coming years. In many instances the reasons for non-approval of off-patent biogenerics is regulatory or legal and not scientific. Although biologics have revolutionised disease treatment, some of these therapies are prohibitively expensive. For instance, treatment with Avastin for colon cancer costs about US$ 100,000 per year per patient and the life-threatening enzyme deficiency Gaucher’s disease treated with Cerezyme, can cost up to a staggering US$ 300,000 per patient for a single year.
Generic biologics offer significant cost advantages and make life saving therapies more affordable to greater number of patients. For example, it is expected that a 20 per cent price reduction for just six off-patent biopharmaceuticals in Europe could save 1.6 billion Euros a year. Based on information from Express Scripts, American consumers could save over US$ 71 billion over ten years, with generic biological products. The push for biogenerics has gained further impetus with recent approval of first product in the US—Omnitrope (Sandoz), similar to branded growth hormone (Genotropin / Pfizer), but not interchangeable. Also several products have been approved as biosimialrs in Europe.
Development considerations
Complexity of biogenerics is exemplified with the controversy surrounding the basic issue of naming such products. Several labels have been used to describe generic biopharmaceuticals including biogenerics, biosimilars, follow-on proteins / biologics and multi-source biopharmaceuticals. The concept of “biosimilars” has gained acceptance in Europe as discussed later in this article. Proteins and other biotechnology-derived products are inherently more complex molecules compared to chemically synthesised drugs.
The complexity is enhanced based on host cell (bacterial vs. mammalian) employed to produce these therapeutics and is related to molecular size, possible conformations, number of subunits and post-translational modifications. In addition, inherent heterogeneity of these macromolecules and levels of impurities and contaminants adds to the complexity. The manufacturing processes used to produce biopharmaceuticals are also complex and lend themselves to the often quoted paradigm of ‘product equals process’. This has been extensively used by branded biopharmaceutical companies to raise an alarm for not approving biogenerics. Their concern is that alternative processes for the same drug could potentially lead to dissimilar products. Typically, more complicated structures such as glycosylated proteins are produced in mammalian cells and the simpler non-glycosylated products use bacterial cells. In addition, some biopharmaceuticals may be chemically modified (PEGylated) to enhance therapeutic outcomes.
Biosimilar product development usually involves several comparative studies performed between the generic and reference products. Development programme for biogenerics should address the following items for demonstrating high assurance of safety and equivalence (biosimilarity) with the corresponding branded biologic:
- •Quality Attributes – Comparability of molecular structures of biogenerics and branded products using a battery of analytical techniques
- •Comparability of biological activity using various in vitro and in vivo tests
- •Comparative preclinical safety and immunogenicity trials
- •Comparative abbreviated pharmacokinetics, pharmacodynamics and clinical studies, including bioequivalence, therapeutic equivalence, safety and immunogenicity
- •Post-approval pharmacovigilance studies and reporting as part of a broad risk management system.
Another aspect of development of biogenerics involves the formulation itself, use of any patented ingredients, stability profile and the primary packaging. In many instances the branded product is packaged along with the delivery system (cartridges, injector pens etc.), which may be protected by additional patents. This is an additional challenge to demonstrate biosimilarity of the drug-device combination with the marketed brand product. Controls on manufacturing and fill-finish operations via Process Analytical Technologies (PAT) will ensure product Quality by Design (QbD) at all steps. As often quoted by leading authorities, the science of developing and approving biogenerics exist today. What is needed is a scientifically rational regulatory and socio-economically favourable legal pathway for their approval and marketing, especially in the United States and Japan.
Tale of two approval pathways
(US vs. EU)
The regulatory bodies in Europe (EMEA, CHMP and EC) have taken a significant lead in laying the pathway for approval of biosimilars. An overarching guideline for biosimilar was issued by EMEA in 2004. Since then several product specific guidelines have been issued for insulins, growth hormones, erythropoietins and colony stimulating factors. These guidelines lay out specific details for clinical and non-clinical requirements for comparing products, including comparability assessment of safety and immunogenicity. The European regulatory authorities have taken a path of balancing cost and safety risk, while considering approval of biosimilars, starting with molecules of moderate complexity. Since issuance of these guidances, several biosimilars have received approval in EU countries, including somatotropins, epoetins (alpha and zeta) and filgrastim.
In the US the pathway for biogenerics / biosimilars has not progressed to the level of its European counterparts. This is partly due to competing business interests at stake and the slow pace of regulatory and legislative reforms. Traditionally new drugs in the US have been approved under Section 505(b)1 of the Food, Drug and Cosmetic (FD&C) Act by the pathway of New Drug Application (NDA). Passage of the Hatch-Waxman amendment in 1984 clearly established a legal authority with the FDA for approving generic drugs under Section 505(j) of the FD&C Act. However, the FDA has deemed that this process is unsuitable for approving generic versions of complex biologics. In addition, several years ago new drugs and some older biologics, such as insulins and growth hormones were reviewed and approved by the Center for Drug Evaluation and Research (CDER) division of the FDA, using the NDA route.
In 1990s the approval of all biologics including rDNA derived products was transferred from CDER to Center for Biologics Evaluation and Research (CBER) at the FDA. This agency approved biologics using a Biological License Application (BLA) route, under the purview of Section 351 of the Public Health Service (PHS) Act. CBER does not have a regulatory mandate for approving generic biologics approved by the BLA route under the PHS Act. In recent years there has been a push towards approving some follow-on biologics, using Section 505(b)2 approach. In this approach the generic company uses safety and efficacy data generated by the innovator in requesting approval of their product. This mechanism is currently available for those biologic drugs that were originally approved by CDER using the NDA route. However, products approved under this mechanism may not be considered interchangeable with the marketed reference product.
The recent approval of growth hormone Omnitrope (Sandoz) by the US FDA falls under this category. However, the FDA has issued comparability guidances in past for biologics undergoing manufacturing, process or site changes implemented by innovator companies. At least one branded company (Biogen) successfully argued with the agency that major manufacturing changes to its originally licensed process for Interferon (Avonex), did not warrant additional clinical and safety trials.
The legislative push for biogenerics in the US is aimed at implementing such comparability standards that are allowed for innovator companies. Due to rising healthcare costs, there is tremendous public pressure to control costs of all therapies, including biologics. US Congress has taken the initiative to create a regulatory pathway for biogenerics, with introduction of “Access to Life Saving Medicines Act” by Representative Waxman and Senator Schumer. This law aims at providing FDA with the authority to approve “follow-on biologics” based on abbreviated clinical studies and extensive preclinical testing and in vitro characterisation. It also provides for significant incentives to generic companies for investing in research and development of such products. For example, it grants market exclusivity based on first approval and not first to file. It also provides tax incentives for performing additional studies to demonstrate interchangeability with the branded product.
The law also addresses several loopholes with the current system of patent listing in Orange Book for conventional small molecule drugs. It calls for the generic company to request relevant patents from the innovator and notify about any potential challenges to those patents. Patents which are not disclosed at the time of this notification may not be enforced. Once this law is enacted, it is expected to provide a clearer understanding of the regulatory pathway for approvals of biogenerics in the US. In the meantime, products approved under the older NDA regime, such as insulin, growth hormones, aprotinin, calcitonin, imigucerase, hyaluronidase may be prime targets for follow-on biologic development in the US. Such products may not be interchangeable with currently marketed formulations.
Other issues with biogenerics yet to be resolved include the use of International Non-Proprietary Names (INNs), legal and patent challenges to biosimilars, strategic business alliances thwarting competition, use of country-specific reference products and timing of data and market exclusivities for the branded product.
While Biotechnology Industry Organization (BIO), a consortium of branded biopharmaceutical companies has raised scientific, technological and safety issues as major concerns for biogenerics, it appears more of a business tactic than scientific. As quoted below by a leading generic company executive, use of comparability protocols intended for branded companies themselves is a viable option for approving biogenerics: “The science to create affordable generic biotech drugs exists today… It is being done every time a brand manufacturer changes a manufacturing process or location and uses comparability to ensure the biotech drug will provide the same safety and efficacy. [B]iotech firms routinely justify process and site changes via comparative studies. For example, if an innovator biotech company seeks changes in process supporting the manufacture of their products, or seeks to change the manufacturing location of a product, comparability is the process by which the amended product is judged to provide the same clinical effect and safety profile”.






