
At the outset, we should challenge the basic dogma of receptor pharmacology, i.e. one ligand, one receptor texture and singular efficacy. There is emerging evidence to the contrary, i.e. one ligand, differential pharmacology and multiple mechanisms of action. GPCR drug hunters and thought leaders have started to accept this paradigm shift and I believe that similar analogies can be drawn for other druggable target classes as well. The plethora of GPCR targets (class A, B, C, frizzled and their respective orphans; IUPHAR reference at http://www.iuphar-db.org/GPCR/ReceptorListForward) is promising but discovery teams must appreciate the evolving concepts and differential in vivo efficacy profiles for ligands (selective for the GPCR of interest) that maybe a result of GPCR oligomerisation, GPCR allosterism and functional selectivity. All of these will be covered in some detail in the following sections. The impact of such evolving concepts can be immense both in terms of screening and finding novel mechanisms for clinically validated targets. As an example, more often than not HTS campaigns for GPCR drug targets are set up with a single readout using recombinant receptors in isolation from their native milieu. While discovery project teams assume and sincerely hope for a carryover pharmacology from the recombinant receptor to the ‘diseased’ receptor, it is to the project team’s benefit if a screening campaign or lead optimisation exercise can incorporate orthogonal screening cascades (incorporate > 1 signalling pathway) and if possible, target human cells / tissue so that any differential pharmacology (due to GPCR oligomers and permissive / restrictive conformation) can be accounted for as early as possible. Another approach that has been gaining steam is to exploit GPCR allostery as a versatile way of fine-tuning the endogenous ligand’s affinity for the target. I advocate for HTS screens that will differentiate between allosterism and orthosterism, both for agonists and antagonists.
GPCR allosterism
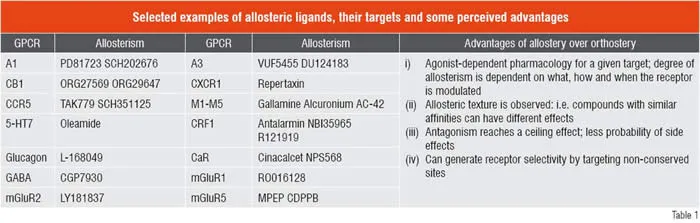
Recent elegant pharmacological studies have identified several GPCRs (muscarinic, adenosine, dopamine, chemokine, CRF1, CaR, mGluRs and GABAB) with allosteric modulatory sites (Gao and Jacobson, 2006; Langmead and Christopoulos, 2006; Milligan and Smith, 2007; Schwartz and Holst, 2007). Several allosteric ligands have nowentered into early development; the marketed drug Cinacalcet is in fact a positive allosteric enhancer for the Calcium-sensing Receptor (CaR) and Maraviroc is a negative allosteric modulator of the CCR5 receptor. As a result, modelling of receptor-ligand interactions has evolved to incorporate allosteric ternary complex models (Christopoulos and Kenakin. 2002; May et. al., 2007). Allosteric modulators (both positive and negative) can have different effects on the endogenous ligand’s affinity and efficacy and hence offer a texture to the pharmacology. Allosteric and orthosteric sites on a GPCR are conformationally linked; it can be argued that GPCRs are naturally allosteric as they do interact with multiple protein subunits and binding of a ligand conformationally selects one over the other. Receptor dimers / higher order oligomers can also be allosterically modulated (allosteric protomer) as was recently demonstrated by Redka et. al., (2008) for the M2 muscarinic receptor. The concept and success of allosterism has opened up the field and brings in immense hype and also genuine promise to feed the GPCR pipeline. From a medicinal chemist’s point of view, it opens up endless possibilities to fine tune a GPCR that may not be restrictive from an IP perspective; i.e. a novel chemotype on a novel GPCR or even on a GPCR that ‘has been around’ for a long time. A unique property of allosteric modulator is their specificity or dependency on the orthosteric agonist and its ability to impart a different texture depending on receptor conformation. This concept can be immensely beneficial in drug discovery and may aid in sparing the physiological roles of GPCR and only modulate the ‘diseased’ receptor, which is conformationally different (for example, a sensitised state of a GPCR that contributes to pain perception).
Another important feature of allosterism is their saturable effect that may translate to less probability of adverse / toxic effects even at full target occupancy and may dissociate duration (function of compound exposure) of action from maximal efficacy (saturable). A big hurdle in allosteric drug discovery is to settle on a screening strategy for a GPCR that is expressed recombinantly in a host cell (conformation ‘A’) in a way that is conformationally different from the GPCR under pathology (conformation ‘B’). In my opinion, it is always advisable to use a native cell system where the receptor is captured in some conformationally restricted state that may mimic the druggable receptor. Because of agonist dependency, it is also critical that allosteric screens use the endogenous agonist where possible, to probe the target of interest. From that perspective, orphan GPCRs may present some operational challenges. There are many GPCR targets, many of which have been clinically validated, that offer a renewed hope for allosteric approach where one may embark on novel chemistry (targets that are chemically intractable for orthosteric ligands) and may eventually improve the therapeutic window and balance the discovery portfolio with low-risk high-reward targets.
Functional selectivity
Although intrinsic efficacy has been viewed as a system-independent pharmacological parameter since the dawn of receptor theory, it is becoming apparent that many ligands (agonists) function differentially and efficacy may swing like a pendulum and it is often difficult to assess the fulcrum in the balance of efficacy-adverse effect profile. Several lines of evidence (reviewed in Mailman, 2007; Urban et. al., 2007) support the notion of functional selectivity:
(i) agonist pairs possess different potency ratios for probing different signalling pathways in vitro that cannot be accounted for methodological differences
(ii) (partial) agonist pairs exhibiting different intrinsic activity ratios (sometimes completely opposite) dependent on the receptor-effector coupling system being tested.
In other words, agonists (full, partial, inverse and ago-allosteric) display functional selectivity or display a protean texture to the agonism. As an example, the antipsychotic aripiprazole (partial D2 agonist) is believed to manifest its efficacy on positive symptoms of schizophrenia (high local dopamine concentrations) by competing with and causing partial antagonism of dopamine stimulus. Contrary to this, when endogenous dopamine levels drop, aripiprazole can then cause partial agonism, highlighting a case of a launched product whose intrinsic activity is dependent on the levels of the endogenous ligand (functionally selective). Whether functional selectivity is a result of one GPCR with many conformation states, GPCR oligomerisation or GPCR-many effector interactions, remain to be elucidated; nonetheless it brings in a lot of promise and hope to find safe and efficacious molecules targeting a GPCR that is otherwise often thought to be a liability for drug development. It is possible to reconcile a scenario where a typical agonist that derives affinity for a single receptor state (and thereby transduce via the same signalling cascade(s) every time the GPCR is turned on) can lead to both efficacy and adverse effects whereas a functionally selective agonist can, in theory at least, with the application of sophisticated pharmacology and medicinal chemistry be tuned to dissociate efficacy from side-effects. Similar concepts can be applied towards antagonists that can also be functionally selective. How can one screen for functional selectivity in early discovery? The answer to this question is to run HTS campaigns or lead optimisation activities with multidimensional pharmacology screens. In other words, there must be a paradigm shift in screening from a single functional assay (example, calcium flux in FLIPR) to other end points that the receptor of interest couples in the host cell being studied (example, MAPK activation or other determinants of cell morphology) which can be translated to pathophysiology.

GPCR oligomerisation
This evolving concept of GPCR oligomers has taken a new turn and several companies have embarked on screening platforms to identify novel chemotypes that targets more than one GPCR unit (CARA Therapeutics, Dimerix Bioscience, PatoBIOS Incorporated). The savvy drug hunter can appreciate that GPCR homo and / or heterodimers can have a significant impact on signalling and eventually on the success of drug discovery projects that often begin with a singular target in mind. For example, GABAB receptor exits as dimers and GABABR2 is required for GABABR1 cell-surface expression and the latter possesses the orthosteric ligand-binding domain whereas the former aids in G-protein coupling. Several other examples of GPCR hetero-dimerisation have recently resurfaced: ?2 adrenergic & prostaglandin EP1 receptor, KOR & DOR (opioids), CB1 & A2A and AT1-B2 (reviewed in Panetta and Greenwood, 2008). Although we appreciate the function of GPCR oligomers (expression, internalisation, signalling via different G proteins), it is often difficult to apply a GPCR oligomeric drug discovery approach because of the unknown receptor mix (monomers or dimers or higher order oligomers) in pathology. It might be more rational to begin with the diseased tissue of interest, assess the GPCR by biochemical means as to whether it exists as monomers or dimers and then reconstitute the receptor pair in cells for screenings that may have a direct impact on the disease itself. The other approach is to choose native cell systems that may express both receptor pairs, as was the case of GPR50-MT1 receptor in a human endothelial cerebral cell line (hCMEC/D3). Whether such efforts will lead to GPCR oligomer selective ligands with therapeutic benefits remains to be seen (Milligan, 2008).
Future of GPCR research
The future of GPCR research is full of optimism and it is upon us, the research community, to tackle the challenges thrown by Mother Nature, expand the pharmacological toolbox and convert new discoveries to medicines. In addition to the three areas of changing paradigms discussed, some other paradigm shifts, although not mentioned in this article should not be forgotten, i.e. role of RGS proteins, G-protein independent GPCR signalling (7-TM receptors), G-protein activation without GPCR activation and the role of accessory proteins in shaping GPCR pharmacology. In the end, GPCR drug discovery should aim at addressing allosteric modulators and also try to address target tissue pharmacology as early in the discovery cascade with different readouts with a view to capture different states of the receptor. From a drug hunter’s perspective, it can be said that the screening strategy is critical to the success of the project: for a given GPCR project it is best to frame a hypothesis, settle on a mechanism (orthosteric versus allosteric; monomers versus dimers) and cast a wide fishing net to capture a broad set of compounds. It is better to have a peripheral vision than a tunnel vision in our ever so challenging business of drug discovery and development.
References
1) Christopoulos A, Kenakin T (2002) Pharmacological Reviews, 54:323
2) Gao Z-G, Jacobson KA (2006) Drug Discovery Today, 11:191
3) Langmead CJ, Christopoulos A (2006) Trends in Pharmacological Sciences, 27:475
4) Mailman RB (2007) Trends in Pharmacological Sciences, 28:390
5) May LT, Leach K, Sexton PM, Christopoulos A (2007) Annual Review of Pharmacology & Toxicology, 47:1
6) Milligan G (2008) British Journal of Pharmacology, 153:S216
7) Milligan G, Smith NJ (2007) Trends in Pharmacological Sciences, 28:615
8) Panetta R, Greenwood MT (2008) Drug Discovery Today, 13:1059
9) Redka DS, Pisterzi LF, Wells JW (2008) Molecular Pharmacology, 74:834
10) Schlyer S, Horuk R (2006) Drug Discovery Today, 11:481
11) Schwartz TW, Holst B (2007) Trends in Pharmacological Sciences, 28:366
12) Urban JD, Clarke WP, Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, Miller KJ, Spedding M, Mailmam RB (2007) The Journal of Pharmacology and Experimental Therapeutics, 320:1





