This article goes beyond published guidance documents of the US Food & Drug Administration (FDA) and outlines the latest trends in its feedback that must be considered when applying human factors engineering (HFE) during combination product development. The authors outline key research and analysis activities and nuanced scoping considerations for the HF validation test required for FDA approval

Although less well known than other drug development phases, human factors engineering (HFE) is critical to meeting regulatory expectations for drug-device combination products. Many markets, including the European Union and China, have standards and guidelines for applying HFE. However, this article focuses on the HFE requirements of the FDA (sometimes referred to hereafter as “the Administration”), which are currently considered to be more rigorous.
HFE is a multi-disciplinary field that involves engineering, psychology and design and is focussed on ensuring that a product’s design is well-matched to the product’s intended users. Specifically, products should be safe, usable and satisfying while accounting for intended users’ skills, knowledge, abilities and limitations.
HFE activities occur at many stages of product development. They can include user research, development of use-related risk documentation, formative evaluations, and a human factors (HF) validation test, also known as a summative usability test, among others. HFE activities help fulfill regulatory requirements associated with product safety and help designers identify specific product requirements, such as an appropriate force required to activate an auto-injector’s drug delivery.
The FDA has established HFE guidelines for drug-device combination products, which include injection devices (e.g., pre-filled syringe, auto-injector, on-body injector), inhalation devices (e.g., inhalers, nebulisers) and other devices (e.g., nasal delivery devices). The Administration is particularly concerned with these products’ usability and use safety, partly because many of these products are used by laypeople, such as patients and caregivers, without medical training. While this article focuses on drug-device combination products, the FDA might also expect HFE to be applied to oral medications and/or drug products with unique treatment regimens and/or packaging.
The FDA expects all materials the user interacts with (i.e., the user interface) will be subject to HFE activities. Per the Administration, “user interface” refers to more than just the physical product, such as a pre-filled syringe’s plunger, stopper, barrel, flanges and needle. This term also includes the packaging, labelling and any instructional materials. When discussing “combination products” or “product,” we are referring to this comprehensive user interface (UI) definition.
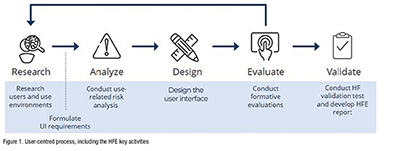
HFE efforts can be subdivided many ways, but we typically distil the process into six activities. Notably, the scope of these activities depends on the intended users, indications for use, associated use-related risks and the ability to leverage prior HFE work. For example, pharmaceutical companies may be able to leverage the platform manufacturers’ early-stage HFE work upon selecting a platform drug-delivery device. (Figure 1).
Research users and use environments
User research activities, including contextual observations, interviews with intended users and literature reviews, help inform the product design, deepen understanding of user needs and determine the intended use environments’ key characteristics. These activities generate insights needed to develop user profiles, use environment descriptions and provide context for the product’s use.
Formulate UI requirements
UI requirements describe components, features and characteristics you consider ‘must haves’ in your final design. These requirements are derived from user research, the use-related risk analysis (URRA) and knowledge about existing products. They describe how the new product should be designed to enable safe and effective use.
Conduct use-related risk analysis
The URRA helps to identify possible use errors and resulting potential harm, such as injury, death and compromised medical care. Using a task analysis, known problems analysis and/or hazard analysis, you can identify more potential use errors, describe the resulting hazardous situation and categorise the severity of the potential resulting harm(s) in an itemised table.
Conduct formative evaluations iteratively
Formative evaluations help assess the usability and use safety of an in-progress design. Examples include an expert review, such as a product critique performed by HFE practitioners and/or designers, a cognitive walkthrough during which an intended user shares feedback while interacting with the product’s prototype and a formative usability test involving simulated product use and related interviews. Formative evaluations are technically optional but can be valuable activities highly encouraged by the FDA.
Conduct HF validation test
The FDA requires an HF validation test to demonstrate that the product is safe and effective for use. Such HF validation testing is needed for new products and marketed products that have been changed in a way that results in new or impacted critical tasks. Typically, the HF validation test simulates actual product use (e.g., injecting into a cushion rather than administering a real injection), a distinct difference from clinical trials. We discuss this pivotal task in more detail below.
Develop HFE report
Assuming favourable HF validation test results, the HFE report asserts that the product is safe and effective to use and provides a comprehensive overview of all HFE activities conducted throughout the product’s development. The FDA provides a detailed outline for this report and covers the intended users, use environments and training, if applicable; known use problems with previous or similar products; the URRA; formative evaluations and any subsequent changes made to the product design; HF validation test method, including any changes implemented based on feedback from the FDA; and HF validation test results and residual risk analysis.
The HF validation test is the HFE version of a pivotal trial and can require rigorous planning, data collection and analysis. The test involves a production-equivalent product and representative users to serve as participants.
Identifying evaluation activities
Evaluation activities are the core of the usability test, occurring after informed consenting and an introduction and before open-ended, debriefing questions. Based on FDA guidance, evaluation activities include all critical tasks. The FDA defines these as “user tasks that, if performed incorrectly or not performed at all, would or could cause harm to the patient or user, where harm is defined to include compromised medical care.” Evaluation activities are both hands-on, simulated product use scenarios and knowledge-based assessments to check participants’ understanding of essential steps that cannot be easily evaluated through observation, such as storage, preparation and disposal. Each scenario should represent actual use, including the use environment and setup.
Defining and recruiting representative users
Per the FDA, an HF validation test requires a minimum of 15 participants per user group. The general expectation is that a single user group will have shared characteristics, including backgrounds, abilities and experience with similar devices. Your product might have one user group (e.g., patients) or many (e.g., adolescent patients, adult patients, lay caregivers and healthcare professionals). Your user research findings and the product’s indications for use inform the type and number of user groups required.
The FDA expects HF validation test participants will represent actual, intended users. However, recruiting people diagnosed with rare medical conditions can be challenging. Recruiting “proxy” (i.e., surrogate) participants with similar backgrounds and characteristics to the target population might be acceptable in these cases. When taking this approach, it is suggested that you thoroughly document your attempts to recruit users and submit a convincing justification regarding why these proxy participants are appropriate. As an added assurance, you can submit your HF validation test protocol to the FDA before conducting the HF validation test to get the Administration’s feedback on the test approach.
Conducting test sessions
Typically, two HFE practitioners conduct each test session in person. One individual administers the session and leads interviews while the other records test data. While many variables affect the duration and scope of a test session, a combination product test session might last one to two hours and usually involves a single participant.
Data collection and reporting
Each task, or use step, should have clear pass/fail criteria, and the HFE practitioners conducting the test should assess participants’ performance. Participant performance on each task should be categorised as one of the following:
Per the FDA, test personnel should perform a root cause analysis to determine why each use error or interaction difficulty occurred by considering a participant-reported root cause and their HFE expertise. As such, the moderator interviews the participant about what led to a particular decision, action or difficulty toward the test session’s end.
The HF validation test report summarises observed test data and describes all use errors, close calls, difficulties and root causes. Rather than relying on quantitative analysis, a product has passed or failed the HF validation test based on reviewing all use errors and interaction difficulties in the context of the URRA. Determining if the HF validation test findings do not pose an unacceptable risk to users (the residual risk analysis) is a key component of the HFE report.
Conducting an HF validation test before a clinical trial can provide added assurance of patient safety and confidence in trial results. If clinical trial participants are not using a product correctly — for example, if a participant lifts an auto-injector from the injection site prematurely — clinical data regarding the drug’s efficacy will be compromised.
When conducting a pre-clinical trial HF validation test, it can reflect the clinical trial conditions. For example, if healthcare professionals (HCPs) will use the product during a clinical trial, the pre-trial HF validation test could include only HCPs as opposed to also including other identified user groups. Nonetheless, the FDA expects HF validation data based on the production-equivalent product, meaning a second (full-scope) HF validation test is likely necessary.
Key takeaways
Scoping and planning for HFE activities can seem daunting. However, with proper execution, HFE work can enable compliance with regulatory expectations, help ensure the product is safe and usable and facilitate a positive user experience, promoting commercial success. To streamline processes and help establish a smooth trajectory of HFE activities, consider the following:
References
Food and Drug Administration. (2016). Applying Human Factors and Usability Engineering to Medical Devices (FDA-2011-D-0469). Retrieved from: https://www.fda.gov/media/80481/download
Food and Drug Administration. (2016). Human factors studies and related clinical study considerations in combination product design and development: Draft guidance for industry and
FDA staff (FDA-2015-D-4848). Retrieved from: https://www.fda.gov/media/96018/download
Food and Drug Administration. (2018). Contents of a complete submission for threshold analyses and human factors submissions to drug and biologic applications (FDA-2018-D-3275). Retrieved from: https://www.fda.gov/media/122971/download
Food and Drug Administration. (2022). Content of human factors information in medical device marketing submissions: Draft guidance for industry and food and drug administration staff (FDA-2015-D-4599). Retrieved from: https://www.fda.gov/media/163694/download

Allison Strochlic has spent nearly 20 years applying HFE principles to medical and pharmaceutical product development. She delivers HFE workshops and advises clients on strategies to HFE to meet the FDA’s and other regulators’ expectations and leads key HF meetings with regulators. Strochlic has undergraduate and graduate degrees in HF, is a Certified HF Professional, and is co-author of the book Usability Testing of Medical Devices.

Yvonne Limpens has 10 years of experience delivering human factors (HF) services to the medical, pharmaceutical, and IVD industries. Limpens delivers HFE workshops, advises on HFE strategy, leads research and analysis activities and develops key deliverables to support FDA and international regulatory submissions. Limpens holds a bachelor’s degree in industrial design and a master’s in human technology interaction.

Katelynn Larson has five years of experience supporting technical, regulatory and other forms of documentation for the insurance and human factors industries. She supports developing assets regarding human factors activities for a variety of medical device and pharmaceutical product manufacturers.













