
Most therapeutic proteins in clinical trials and on the market are to a variable extent immunogenic. Formation of anti-drug antibodies poses a risk that should be assessed during drug development, as it possibly compromises drug safety and alters drug characteristics including pharmacokinetics and bioavailability.
The immunogenicity risk assessment is dependent on the nature of the protein therapeutic, and should be analysed on a case-to-case basis.
Immunogenicity
The immune system has evolved to protect hosts against potentially harmful antigens. Immunogenicity is the immune response a host mounts against an antigen, such as a protein therapeutic. Typically, immunogenicity is characterised by measuring the production of Anti-Drug Antibodies (ADA) against the protein therapeutic.
The fast growing number of therapeutic proteins and related immunogenicity data shows that most of them induce ADA. These ADA can have an impact on drug-characteristics including the pharmacokinetics, bioavailability and drug clearance rate.
While in many cases the ADA are non-neutralising antibodies, there are documented cases where the immunogenicity gives rise to Neutralising Antibodies (NAb). These NAbs have a direct effect on the drugs’ effector-function. Upon development of a protein therapeutic, the likelihood of observing immunogenicity has to be estimated. Moreover, the severity of an observed immunogenicity should also be evaluated. The severity of this response has to be evaluated on a case-to-case basis, as the immune response ranges from a transient appearance of ADA with no clinical effect to severe life-threatening conditions. Both industry and regulatory instances are currently developing immunogenicity risk assessment strategies for protein therapeutics.
Characterising immunogenicity
The measurement and characterisation of antibodies formed against a therapeutic protein by a host is not without technical challenges. A stratified approach (Figure 2) includes several assays to be optimised and validated for each individual drug, in order to
1. screen for circulating antibodies against the protein therapeutic
2. to confirm the positive read-outs with a competitive immunoassay, thereby differentiating false positive read-outs from the actual positive serum samples
3. characterise the type of response and
4. screen for NAbs
Interpretation of immunogenicity percentages’ data for protein therapeutics should be done with care. The measured immunogenicity is to a certain extent dependent on the assays used, and moreover, many of the studies comprise only a small number of patients, sometimes submitted to different treatment schemes based on their individual medical history and disease state.
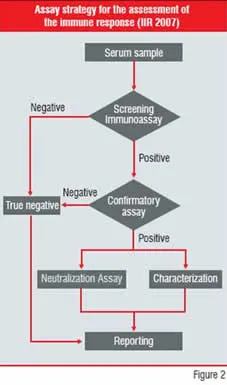
Figure-2: Assay Strategy for the
assessment of the immune response
(IIR 2007) An example of the variability of observed immunogenicity is Alemtuzumab, a humanised anti-CD52 antibody. The observed ADA response in the treatment of rheumatoid arthritis varied between 0 to 75 percent throughout six clinical studies published between 1995 and 2005. The combined data resulted in an immunogenicity of 45 percent for 120 patients. On the other hand, in the treatment of patients with B-cell Chronic Lymphocytic Leukemia (B-CLL) only 1.9 percent ADA response was measured for a group of 167 subjects, suggesting a possible influence of the disease state on immunogenicity.
Similarly, Rituximab, a chimeric antibody directed against CD20, does not elicit ADA when used to treat patients suffering from B-CLL. This again may be explained by the antibody causing B-cell depletion, the presence of a B-cell lymphoma and the concomitant use of immunosuppressive drugs, three factors that hamper the overall production of antibodies. Rituximab administered to patients with auto-immune disease like systemic lupus erythomatosus or primary Sjogren’s syndrome showed 65 and 27 percent immunogenicity respectively.
Pre-clinical immunogenicity assessment
For the time being, there are no in vitro methods available to measure the antibody responses raised against protein therapeutics.
The HLA-peptide complex is then transported to the APC surface, where the complex can be recognised by T-cell receptors. This will then cascade the production of cytokines triggering the proliferation and activation of B-cells producing the antibodies against the protein.The HLA-peptide complex is then transported to the APC surface, where the complex can be recognised by T-cell receptors. This will then cascade the production of cytokines triggering the proliferation and activation of B-cells producing the antibodies against the protein.The HLA-peptide complex is then transported to the APC surface, where the complex can be recognised by T-cell receptors. This will then cascade the production of cytokines triggering the proliferation and activation of B-cells producing the antibodies against the protein.The HLA-peptide complex is then transported to the APC surface, where the complex can be recognised by T-cell receptors. This will then cascade the production of cytokines triggering the proliferation and activation of B-cells producing the antibodies against the protein.
Pre-clinical immunogenicity assessment
For the time being, there are no in vitro methods available to measure the antibody responses raised against protein therapeutics.
Therefore, one has to rely on other approaches to estimate the expected immunogenicity of a protein therapeutic. One possibility is to estimate the protein therapeutics’ T-cell epitope content by computer based methods, or by in vitro measuring the level of T-cell activation upon administration of the protein therapeutic to donors or patient material.
Th-epitopes
Being only a part of the host’s immune system, the mechanism to generate antibodies against the protein therapeutic or any other antigen involves at least three cell types:
i) B-cells producing antibodies directed against the antigen
ii) T-helper cells supporting this function by the production of cytokines, and
iii) Antigen Presenting Cells (APC) stimulating the T-helper cells. Typical APCs include dendritic cells, macrophages and B-cells
The APCs take up protein through endocytosis, cleave the protein into peptides in the endosomes, where the peptides can be loaded on membrane-bound Major Histocompatibility Complex (MHC)—in humans these are called Human Leukocyte Antigen (HLA)—class II receptors (Figure 1).
The HLA-peptide complex is then transported to the APC surface, where the complex can be recognised by T-cell receptors. This will then cascade the production of cytokines triggering the proliferation and activation of B-cells producing the antibodies against the protein.
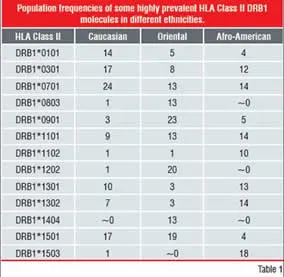
Table-1: Population Frequencies of some highly
prevalent HLA Class II DRB1 Molecules in
different ethnicities HLA polymorphism
As the cascade-process is critically dependent on the presence of peptides that can bind to the HLA Class-II receptors, one could minimise the immunogenicity of a protein therapeutic by selecting protein therapeutics with as little T-cell epitopes as possible.
However, the HLA Class-II system contains many polymorphisms, in order to be able to respond to a broad range of “pathogens”, translating into several HLA receptor allotypes on the cell surface, each with different peptide-specificity.
The diversity is generated:
(i) by the presence of several HLA class-II genes, and
(ii) by a very high degree of polymorphism of most of these genes (Table 1)
Therefore, assessment of the immunogenicity of a therapeutic protein should take into account the prevalence of the different variants within the patient population that is being targeted.
In silico approaches
As peptides bind on HLA in an outstretched fashion (Figure 1), this greatly limits the number of possible binding modes of any peptide to the receptors, as well as the number of interactions between its side-chains. This allows building models that predict the affinity of a peptide for a particular HLA allotype.
The previous generation tools to predict binding peptides to HLA merely focussed on sequence comparison in observed binding peptides. The earliest such models were statistical. Based on a number of experimentally known epitopes, the binding peptides can be aligned to look for amino-acid preferences throughout the sequence positions. Given the final alignment, a statistical profile or matrix is then constructed for the binding groove positions and any new peptide can be aligned to that. The alignment score of the peptide against the matrix is then a measure of its affinity. Rankpep, Propred, Tepitope, and Syphpeiti are examples of such statistical methods. More elaborate models based solely on inference have been constructed as well, using neural networks and classification trees.
Structure-based methods are more recent. These directly model the molecular interactions between a peptide and receptor, using force-fields. This has the advantage that there is less possibility of overfitting the model towards experimental data, which is a serious problem in statistical methods. Structure-based approaches such as Epibase can predict peptide affinities for any HLA allotype, provided that a good model of its structure can be created. For most allotypes this is feasible given careful homology modelling.
The accuracy of predictive methods has evolved strongly over the past 15 years. In Figure 3, the high accuracy of structure-based methodologies is illustrated by comparing three methods, Rankpep, Propred and Epibase, on six of the major HLAII receptors.
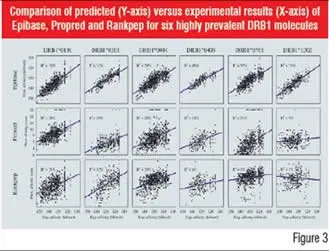
Figure-3: Comparison of predicted (Y-axis) versus
experimental results (X-axis) of Epibase, Propred and
Rankpep for six highly prevalent DRB1 molecules In vitro approaches
While in silico methods assist the R&D process in selecting the lowest immunogenic lead-candidates, they can be useful in some applications to measure the actual T-cell activation level on donor or patient material. Indeed, when comparing different formulations of a drug, or when comparing a biosimilar product with the reference product, the T-cell activation and proliferation assays can document the comparability and immunogenicity risk.
A number of assays have been developed to characterise the T-cell responsiveness. Typically Peripheral Blood Mononuclear Cells (PBMC) from patients or naïve community donors are harvested and primed with the protein therapeutic. After a number of days of culture, the cells are restimulated with autologous PBMC and the protein or derived peptides. The T-cell stimulation can then be determined by a suitable proliferation assay. Routinely, Enzyme-linked Immunospot analysis is used to measure the number of cytokine secreting T-cells. By using Fluorescence Activated Cell Sorting (FACS) based systems, the read-outs can characterise the specific T-cell subsets that are being stimulated, thereby refining the interpretation of the type of response triggered. Typically, population wide responses have to be assessed, and therefore the in vitro assays are performed on 50 or more donors.
Conclusions
In silico T-cell epitope characterisation can reliably guide experimental analysis, thereby significantly reducing cost as well as enriching data interpretation of the in vitro T-cell activation data at the HLA allotype level. A combined approach is essential to address important yet poorly understood issues in immunogenicity, such as the immunodominance of T-cell epitopes.
The combined technology allows to estimate the expected immunogenicity of a protein therapeutic. Since the actual measurement of ADA is only possible during clinical trials, the pre-clinical T-cell read-outs allow optimisation of the lead selection of the formulation process and support a comparability analysis. As the animal models for immunogenicity testing are low to non-predictive for the immunogenicity observed in humans, the T-cell work forms an alternative assessment method prior to the first dose in humans.
References
- EMEA “Guideline on immunogenicity assessment of Biotechnology-derived therapeutic proteins” 2007, EMEA/CHMP/BMWP/14327/2006 Draft dd. Jan 24 2007
- IIR “Immunogenicity of Biologics”, Prague 2007
- SHANKAR G, RENDLEY C, STEIN KE“A risk-based bioanalytical strategy for the assessment of antibody immune responses against biological drugs “ Nature VAN WALLE I, GANSEMANS Y, PARREN PWHI, STAS P, LASTERS I “Immunogenicity screening in protein drug development” Expert Opinions in Biological Therapeutics (2007) 7:405-418





