
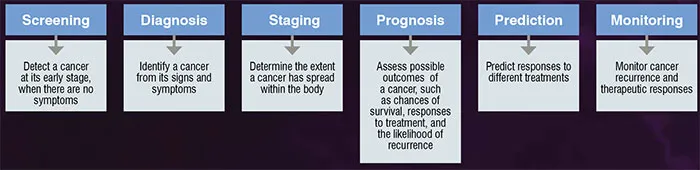
Biomarkers or biological markers cover a wide range of substances that can be measured from body tissues, cells, blood or fluid. For instance, a cell expresses genes when they are required for biological processes, and the measurement of gene expressions under different physiological conditions provide essential clues of gene functions. Therefore, biomarkers play essential roles in understanding complex biological mechanisms, as well as in diagnosis, prognosis and treatment of diseases, such as cancer. The desirable characteristics for ideal cancer biomarkers are non-invasive, low cost, simple to perform, discriminative, informative and produce high accuracy, sensitivity and specificity in clinical applications. Nevertheless, having these ideal characteristics remain as challenges in cancer biomarker discovery. Low specificity is the situation when a test yields false-positive results, causing unnecessary anxiety and treatment to a patient. While, low sensitivity is the situation when a test yields false-negative results, which cause a false sense of security to a patient. Figure 1 shows the possible applications of cancer biomarkers and their respective role.
Biomarker discovery is the process of identifying and measuring the intrinsic features of high-throughput molecular profiling data, such as microarray data, or also known as gene expression data. Microarray data analysis is a powerful preclinical exploratory study for discovering potentially useful biomarkers. A microarray is an ordered collection of biological materials printed onto a small solid substrate such as membrane and glass slide. The common type of array is DNA microarray, which is a glass slide with thousands of spots or probes. Each fixed spot contains identical DNA molecules that correspond to a gene. Microarray data is used to analyse gene expressions within a single sample or to compare gene expressions in two different cell types or tissue samples, such as between healthy (normal condition) and diseased tissues (test condition). Small in size with a large number of genes, microarray has become an indispensable tool to assay the expression levels up to thousands of genes simultaneously in a single experiment.
Some of the well-known microarray platforms include AffymetrixGeneChip, Agilent, and Illumina Bead Chip. Although a microarray’s platform design is subject to include only known genes, it is still considered less biased compared to other high-throughput technologies. Besides, it could provide hints about functional relationships and interactions among genes. Its sensitivity allows detection on very low expressions of mRNA. Examples of microarray-based biomarkers that have been approved for clinical tests include MammaPrint, Roche AmpliChip, Rotterdam Signature, ColoPrint, and NuvoSelect. Despite these promising applications, microarray-based biomarkers are not widely used by either organisation issuing clinical guidelines or expert panels. Remaining challenges that need to be addressed in microarray data analysis are such as cost of development, false-positive errors, data quality, data qualification and interpretation. Besides, lacking organised and integrated resources have worsened the situation.
Data mining provides a wide range of methodologies and tools in microarray data analysis to uncover novel cancer biomarkers and to understand the underlying genetic causes of cancer diseases. Its capability to cope with high-dimensional data make it more preferable compared to conventional statistical methods. Typical microarray data analysis with data mining techniques includes gene selection, classification and association rule mining. The main goal of microarray data classification is to build an efficient and effective classifier that is capable of differentiating gene expression profiles for accurate disease diagnosis or prediction. However, due to a large number of genes and small samples size, traditional statistical and classification techniques are not able to deal with it efficiently, leading to false-positive and overfitting problems, as well as reducing the accuracy and speed of classifiers. Therefore, gene selection, or also known as feature selection, is an essential task for microarray data classification to identify differentially expressed genes and to reduce dimensionality by removing irrelevant, redundant and noisy data. The k-nearest neighbours, naïve bayes, logistic regression and support vector machine are popular classification algorithms used for microarray data classification. These classifiers have shown good performances when combined with suitable gene selection. Figure 2 presents the generic flow for microarray data classification.

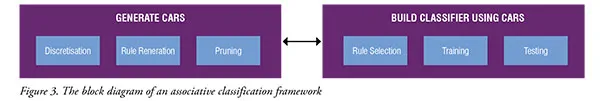
Along with that, Associative Classification (AC) has gained popularity in microarray studies to classify data and to uncover to discover interesting biological relationships from large microarray datasets. Such information is indeed important to extract relevant gene markers, which in turn can create more reliable and accurate cancer diagnosis. AC is a hybrid approach that integrates both classification and Association Rule Mining (ARM). In data mining, ARM is also known as association analysis or frequent pattern mining. ARM is widely used in businesses to predict customer purchasing behaviours called market basket analysis. It works by analysing customer transactions to identify frequently co-occurred items. The same idea can be applied to analyse the human genome, which contains about 20,000 to 30,000 of genes as interacting items. Classification based on associations approach aims to identify a subset of rules, known as Class Associative Rules (CARs), whose consequent are restricted only to target class labels. The classifiers built by existing ACs had proven to produce better accuracy and could improve the understandability and reasoning of a problem. With the assumption that a good classification resulted from a good biomarker identification or the other way round, our study attempts to achieve this goal by using the AC approach. The common strategy in AC is to decompose a problem into two major tasks, as shown in Figure 3. The first task is to generate a complete set of CARs that satisfy userdefined values, such as minimum support and minimum confidence. The second task is to build a classifier based on the strongest CARs
The problem of mining microarray data with AC can be described more formally. Let T = {t1, t2, …, tm} be a dataset or transaction database that contains all transactions. Let G = {g1, g2, …, gn} be the set of all items found in T, and let C = {c1, c2, …, ck} be the set of class labels. Each transaction ti consists of a set of items X, which has been labelled with a class y, such that X G and y C. In other words, a transaction represents a set of expressed genes for a sample or patient, and a transaction database includes all gene expressions recorded from a microarray experiment. Below are the definitions for some of the terms in AC:
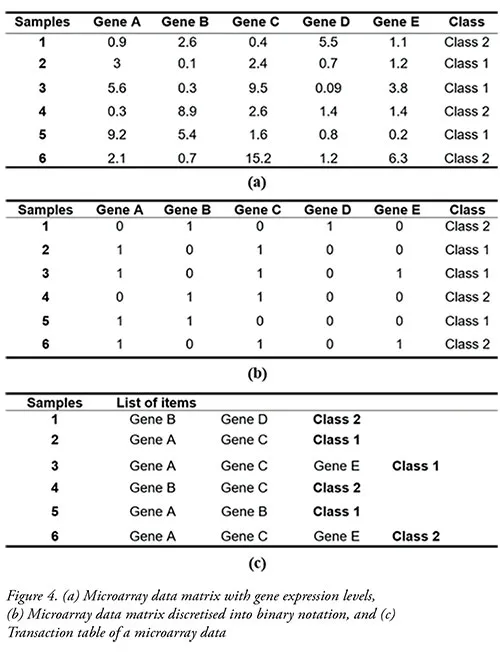
• Transaction database:Figure 4 (a) illustrates an example of a microarray data matrix with relative gene expression values. The genes are treated as items, while the array of samples are treated as transactions. As the microarray data consists of continuous attributes, it needs to be discretised and treated as categorical attributes. Figure 4 (b) shows an example of microarray data that had been discretised into a binary matrix. In discretisation, a certain cutoff value is applied, where an expression value that is above the cutoff value is considered highly expressed and is assigned a value of 1. Otherwise, it is considered highly repressed and is assigned a value of 0. Discretised microarray data matrix can be further transformed to a transaction table, as shown in Figure 4 (c).
- Item: An item is an attribute-value pair of the form (gi, v), where gi G is an attribute taking a value, v, such as an expression value. In certain cases, an attribute can also take multiple values.
- Itemset: An itemsetX, where X G is a set of zero or more items. A k-itemset is an itemset that contains k items, such as {Gene A, Gene C, Gene D} is a 3-itemset.
- Class Associative Rule: A class associative rule is an implication expression of form X y, where X G, y C, and G C = . The left-hand side itemset is known as the antecedent, while the right-hand side itemset is known as the consequent. The consequent of a CAR must be an itemset with a single item from the class label of set C. X and y are non-overlapping item sets.
- Support: The support of a CAR, X->y is defined as the percentage of transactions in the database that contain X and is associated with class y, which is supp(X Y) = count(X y) / count(X). Support is an important indicator to show the frequency of occurrence for a rule. Non-frequent rules that did not satisfy user-defined minimum support can be pruned out as their occurrence could be simply due to chance.
- Confidence: The confidence of a CAR, X->y is the percentage of transactions in database that contain X also contain class y, which is conf(X Y) = count(X Y) / count X). Confidence indicates the predictability and reliability for a rule. Therefore, rules that did not satisfy user-defined minimum confidence can be pruned out.
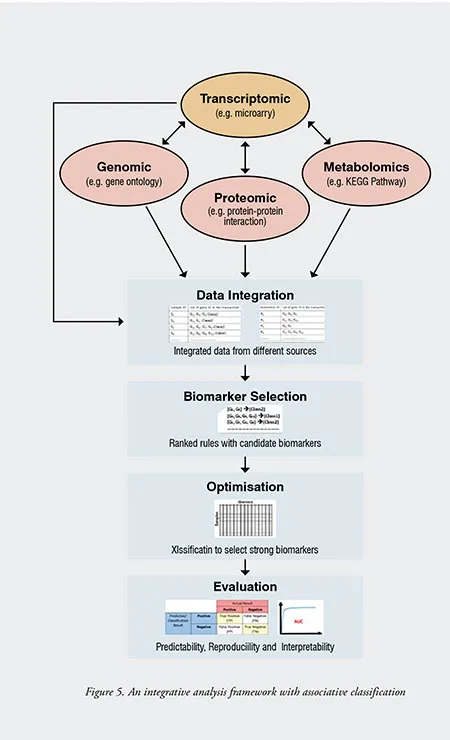
In the past few years, there are also efforts directed toward knowledge-driven or integrative analysis, which combines microarray data with other data sources, such as prior biological knowledge. An integrative analysis in data mining means to combine heterogeneous data, information and knowledge for the generation of higher-level knowledge and new testable hypotheses. An integrated system of multiple types of high-throughput functional genomic data is expected to facilitate fast, accurate, systematic identification and prediction of highly complex biological data. To achieve that, sophisticated computational and analytical methods are needed to overcome current challenges by increasing the sensitivity and specificity in high-throughput data. Diverse genomic data such as gene ontologies, protein-protein interactions and KEGG pathways are integrated by computational methods to create new integrated data with functional relationships between genes. These integrated data can then be used for microarray data classification, and evaluation is done using cross-validation or using a test set of labelled data.
Figure 5 presents the proposed integrative analysis framework for the integration of heterogeneous biological data (e.g. transcriptomic, genomic, proteomic, and metabolomics data), with the processing components of data integration, biomarker selection, optimisation, and evaluation. An associative classification algorithm is adopted to generate the desired number of association rules with the highest support that meet minimum confidence. Several modifications are introduced for mining informative association rules from both microarray and biological transaction tables. With the framework, the top-k CARs generated from different target classes of microarray transaction tables were integrated, and a new ranking algorithm was applied to select a set of strong rules that contain potential biomarker genes that can discriminate classes. An interestingness measurement is proposed to rank the CARs, where the interestingness for a rule is the sum of the information-content from three observations, namely the average information gain, average classification accuracy and modified enrichment score. The most top-ranked class associative rule is considered the most informative rule with the lowest interestingness score, and its genes are considered the most informative genes. Gene subsets are generated from the informative genes of the top-ranked rules, and only the most informative gene subset inputted for the training of classifiers. The best gene subset is the set of genes that can achieve the highest predictive accuracy with less number of genes. The evaluation of the selected biomarkers is based on their predictability, reproducibility and interpretability performances. Also, results obtained were evaluated and compared with other existing methods to determine whether the research problem is resolved or not.
The proposed framework had been tested on four UCI datasets and eight microarray datasets of colorectal and breast cancers. In comparison with other existing methods, it outperformed in terms of classification accuracy and Area Under the Curve (AUC), as well as showing significant reproducibility and interpretability results. These promising results have proven that the proposed method is capable of identifying potential genes, which can be further investigated as biomarkers for specific cancer diseases. The experimental results can be found in the paper Informative top-k class associative rule for cancer biomarker discovery on microarray data. For future works, multi-platform microarray data can be integrated into the same integrative analysis to produce more reliable and accurate biomarker discovery. Moreover, an improved AC method can be introduced to increase the efficiency of rule mining.





