
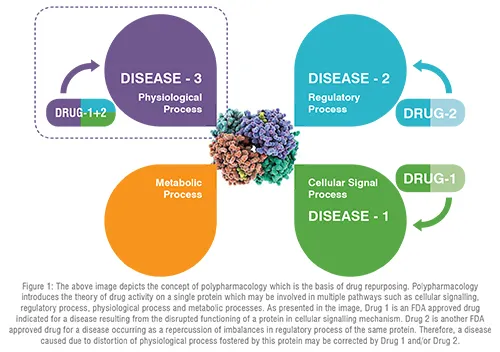
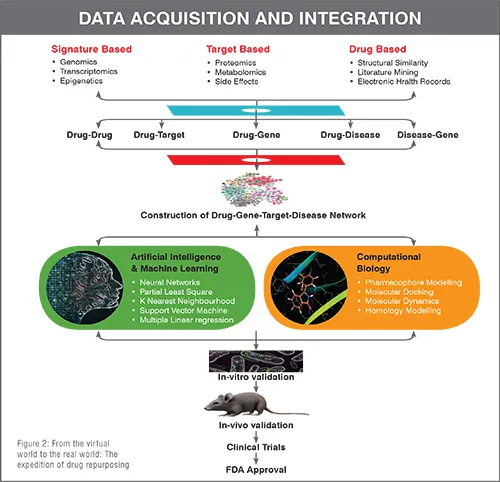
Conventional drug discovery path is arduous, time-consuming, and demands massive fiscal investments. Despite all the efforts put forth during gross drug development processes, newer drug molecules entering clinical trials either lack therapeutic efficacy or demonstrate erratic safety profiles that result in dwindling approval rates for new drug applications and revocation from the market, if already in use. Furthermore, emergence of new diseases along with growing population, dearth of promising therapeutic regimens for neglected tropical diseases and orphan disorders, pandemic, and epidemic infectious outbreaks combined with accelerated drug-resistance, tachyphylaxis, adverse effects, and idiosyncratic reactions are immensely pressurising pharmaceutical companies to design, discover and bring about authentic, safe, and effective drugs to the market in a prudential manner. To cater these never ending therapeutic requirements, a paradigm shift towards drug repurposing (DR), which is deep-rooted in polypharmacology background, is of paramount importance (Figure 1). DR, a present day trend to identify new indications for pre-approved or banned drugs, furnishes an equivalent or superior outcome over the conventional de novo methods. This conspicuous strategy has achieved notable impetus over the last decade. The evolution of DR over years from its humble serendipitous start to the space-age incorporation of artificial intelligence is an enticing feat. Recent updates on multifaceted approaches for DR and the work flow from a conceptual note to approval from regulatory agencies are depicted in Figure 2.
From genes to repurposed drugs, the story of genomics
Genomics is a fascinating omics science that entwines structural and functional genomics with genetic elements. Excavating the gene expressions using genomic insights offer colossal support for discovering novel therapeutic disease targets for DR.
Stephen Wan Leung et al., mined Cancer Cell Line Encyclopedia and The Cancer Genome Atlas data and predicted over-expression of HMGA2 (High Mobility Group AT-Hook 2) in Colorectal Cancer(CC). Subsequent microarray profiling of gene signatures exposed the druggable vulnerability of S100 calcium-binding protein A4 (S100A4) corresponding to the aforesaid gene. Later Connectivity Map (CMap) analysis revealed inhibition of S100A4 can reverse the expression of HMGA2. This analysis identified Niclosamide, an anti-helminthic drug, as a potential suppressor of S100A4 for CC by intervening WNT signaling pathways, which was later validated through in-vitro and in-vivo analysis.
Protein structures to reveal novel treatment options through proteomics
Proteomics portrays the total protein content of a cell, tissue, or organism and investigates protein-protein and protein-nucleic acid interactions that play a key role in regulating protein functions. They provide crucial data on role of a single protein in multiple pathways that might trigger multiple signalling mechanisms which might be involved in disease pathogenesis and side effects of drugs. These complex details reinforce the rationale for implementing proteomic approaches in DR.
Pei-Feng Liu et al., retrieved 1312 approved drugs from the US Food & Drug Administration (FDA) via virtual screening against ATG4B, an autophagic protein. These drugs were clustered and subjected to molecular dynamics (MD) and binding energy calculations. This resulted in 22 drugs, of which, Tolfenamic acid, Mefenamic acid, Ticonazole, and Entacapone were found to inhibit ATG4. Ticonazole was reported to possess the highest inhibition. Further, in-vitro evaluation of Ticonazole also exhibited diminished autophagic activity, cell viability and synergised the cytotoxic effects of starvation in cancer cells. Ticonazole-treated mice in-vivo reduced the xenograft tumour volumes.
Novel drug targets, a gift of gene expression conveyed through Transcriptomics
Transcriptomics describes RNA transcription by genome and illuminates the responses of individual genome against functional and environmental perturbations. This is essential for elucidating the complex mechanisms underlying disease pathogenesis, thereby, assisting in identification of potential disease targets. This approach has paved way for DR research.
Nasir Mirza et al., retrieved transcriptomic signatures of chronic temporal lobe epilepsy from a mouse model and used it as query in ‘The Library of Integrated Network-Based Cellular Signatures’ to identify drugs capable of reversing these signatures. The search resulted in 123 compounds, of which, 36 were shortlisted for Drug set Enrichment Analysis. This identified five drugs that share similar pathways. Sitagliptin, a well-tolerated anti-diabetic drug was one amongst the five. Ultimately, in-vivo validation of Sitagliptin exhibited antiepileptic activity at 500 mg/kg dose.
Uncovering the realm of novel therapeutic possibilities by the assistance of epigenomics
Epigenomics evaluates the induction of abnormal alterations in the phenotypic characteristics by external factors (age, lifestyle and environment or disease state) via DNA methylation, non-coding RNA associated gene silencing, and/or his tone modification, without genetic sequence mutations. This is emerging as an unconventional path for DR.
Paulami Chatterjee et al., obtained 54 Alzheimer’s Disease (AD) related epigenetic genes from Human Protein Reference Database, BioGRID and MENTHA. A curated list of genes was assessed for Epigenetic Protein-Protein Interactions (PPI) and significant proteins extracted from PPI were used to retrieve drugs from Drugbank to construct Drug-Target Network. This resulted in 1,920 drug-target interactions between 886 drugs and 419 proteins. Ultimately, Aspirin, Tamoxifen, Caffeine, Sorafenib, Glyburide, Spironolactone, Methotrexate, Diclofenac, Lamivudine, Ibuprofen and Etoposide were identified as repurposable drugs.
Metabolomics: Using cellular metabolites to unveil therapeutic advances
Metabolomics involves commissioning of sophisticated instrumentation techniques to determine alterations in the concentration of metabolites or metabolic profiles for individual patients. This data on aberrant pathways offers clues to explore novel targets to redirect existing drugs towards DR.
BesteTuranli et al., retrieved 8558 genes overlapped between transcriptomic and proteomic data, 3328 proteome-specific and 2892 transcriptome-specific genes from NCBI GDC and Human Protein Atlas for Genome-scale Metabolic Model creation. Network analysis was carried out with Integrative Network Inference for Tissues algorithm by exploiting iCancer model to identify metabolic reactions specific to Prostate Cancer (PC). This resulted in 2655 genes, 6718 reactions, 86 up regulated and 76 down regulated PC metabolites. Amongst the total, 23 genes were involved in steroid biosynthesis pathways. Subsequently, network mapping via Differential Rank Conservation analysis revealed dominance of lipid and riboflavin metabolism, pentose phosphate pathway, and thyroid cancer in tumour samples. Later, CMap2 was employed to investigate 81 repurposable drugs. Ultimately, Sulfamethoxypyridazine, Azlocillin, Hydroflumethiazide, and Ifenprodil were found to possess significant potential to ameliorate PC.
Structural Similarity: Know the structure and unveil the trait
This approach assumes structurally similar drugs to possess similar pharmacological actions. The hypothesis thus generated focuses on forecasting plausible opportunities to carryout DR research. Various computational approaches like 3D fingerprints, pharmacophore similarity, scaffold similarity etc., have been developed to explore potentially repurposable candidates.
Nikhil Pathak et al., screened the compound library comprising 187,740 from ZINC database against four dengue NS3 proteases. Top 3000 compounds were ranked by considering binding energies and pose scores. Based on interaction profiles, pharmacophore anchors were modelled and core pharmacophore anchors were identified by aligning all pharmacophore anchors for each NS3 protease. This protocol was validated by performing molecular docking with known respective inhibitors against each NS3 protease. Finally, interaction energies were analysed for each anchor and top 100 docking poses were integrated to screen 1384 FDA approved drugs. This yielded Boceprevir, Telaprevir and Asunaprevir which were further evaluated in-vitro using Huh7 and DENV2-NGC virus treated Huh-7 cells for evaluating cytotoxicity and dengue plaque formation. This proved theanti-dengue effect of Asunaprevir.
Electronic Health Records: A massive collection of patients’ data directed towards innovative DR

Real world data on a patient’s encounter with any health care delivery system is documented in electronic health records (EHR). It accumulates patient’s demographics, past medical and medication history, family history, diagnosis, laboratory data along with therapeutic regimens. This common data when extracted using relevant retrieving strategies like natural language processing (NLP) technique shall appraise the unstructured data stored in EHR. Further, this technique if streamlined towards identifying new drug indications serendipitously, serves as an unparalleled approach towardsDR.
Hyojung Paik et al., extracted EHR of 530 K patients and 8693 K drug prescriptions from a tertiary hospital. A novel algorithm “Clinical and Genomics signature-based prediction for Drug Repositioning” was constructed by calculating drug-drug similarity and disease-disease similarity matrices, using Wilcoxon sum test, before and after drug administration. Single similarity matrix was designed by incorporating drug-drug matrix and disease-disease matrix based on their Gene Ontology and PPI. This approach revealed similarity between structures of Terbutaline and Ursodeoxycholic acid (UDCA); Kawasaki syndrome and Amyotrophic Lateral Sclerosis (ALS). EHR also revealed that UDCA and Terbutaline sulfate can be used to treat Kawasaki syndrome and ALS respectively.
Literature-based DR: Mining through history to avail resources for the future
Retrieval of information on disease specific targets buried in enormous valuable literature sources is a herculean task which can be simplified through application of computational techniques to abridge drug discovery timelines.Literature mining (LM) is one such approach that uncovers key elements obscured in textual data resources to frame a substantial research hypothesis. LM is useful in (i) extracting data of research interest from huge literature, (ii)format unstructured data to a well organised one, and (iii) decode the data in an analysable pattern. Currently, numerous hypotheses were generated using LM to promote advanced research in drug discovery and DR.
Giup Jang et al., screened 1,454,763 abstracts from the PubMed database to extract drug-gene co-occurrence and phenotype-gene co-occurrence. Sentences were then parsed to identify entities i.e. genes, phenotypes and drugs. A dependency graph was formulated to extract relationships between entities. Subsequently, Gene Regulation Score (GRS) was calculated to identify significant gene-drug and gene-phenotype relations. Using GRS, Therapeutic Possibility was computed to derive drug-phenotype relationships. The resultant hypothesis elucidated DR potential of (i) Fenofibrate, Losartan, Valsartan and Spironolactone for prostate cancer; (ii) Cimetidine, Dacarbazine, Ezetimibe, Flurbiprofen and Lovastatin for Myelogenous Leukemia; (iii) Chloroquine, Estradiol, Metformin and Propofol for Melanoma; (iv) Sulindac, Tadalafil, Metoprolol and Niclosamide for Bladder cancer; and (v) Chloroquine, Naltrexone and Ritonavir for Cerebro-vascular disease.
The revolutionary application of side effects data to uplift DR hypothesis
Side effect databases provide vast data of undesirable and untoward extensions of drugs’ activity. These unwelcome phenotypes of the drug rely on polypharmacology concept which highlights the likelihood of multi target actions of drugs that are interlaced in numerous disease networks. The utility of this novel conception resulted in the advent of a distinctive DR approach.
Hao Ye et al., mapped 6495 side effects of 2183 FDA approved drugs from SIDER, MedDRA, Meyler’s Side Effects of Drugs and Side Effects of Drugs Annals. Consequently, unique side effect fingerprint for individual drugs was created and similarities among side effects and drug was calculated using Jaccard Index (cutoff = 0.275) in order to construct a drug network. This revealed 17,400 drug-drug pairs, spanning 1647 drugs based on side effect similarity. Of the total, 1234 drugs were selected as they contained a minimum of 2 neighbors which were mapped to 81 Anatomical and Therapeutical Classification (ATC) with 584 unique indications. Dynastat (Parecoxib) initially approved for pain management was found to be enriched in rheumatoid arthritis. Tramadol (analgesic) and Tolcapone (a FDA approved drug for adjuctive therapy of parkinson’s disease) were found to possess anti-depressant activity.
Collaborating approaches for DR: Does strength come from unity?
Aforementioned computational approaches adopted in DR research offer quick forecast, yet, they are not devoid of certain boundaries. In case of target-based approaches, investigation of target binding site is of prime importance in exploring new indications. However, none of the existing algorithms are precise in predicting the binding sites of new or unrelated proteins, thereby restricting their applicability in identification of novel compounds through DR.
Explicating the mechanisms underlying gene expressional changes, post drug treatment, faces challenges in terms of noise that can result in biased network predictions. The same pitfalls are experienced in PPI network construction and mining side effect data.
Drug based approaches, by default, encounters some drawbacks due to deficit of pertinent details related to proprietary norms and erroneous chemical structures selection.
In case of molecular docking studies, ligand matching with binding grove of the target protein is unreliable. Though this technique offers flexibility to the ligand, the receptor is left rigid. This necessitates the exploitation of MD studies which can contribute flexibility to ligand and the receptors, to simulate natural condition, which demands expensive high-performance computing facilities. In order to overcome the inadequacies pertaining to individual approaches, a multifaceted integrative approach is highly recommended to accomplish a vibrant DR outcome.
Ming Zhang et al., developed an innovative DR approach by integrating genomic, proteomic, epigenetic and metabolomic data from Genome Wide Association Studies catalogue, Uniprot, Literature and Human Metabolome Database respectively. 220 genes associated with 244 variants, 98 proteins, 86 metabolites and 14 epigenetic associations were mapped. PPI analysis revealed association of 524 proteins in AD pathogenesis. These proteins were later mapped to FDA approved drugs from Therapeutic Target Database and Drugbank to identify potential repurposable drugs. Amidst the 524 proteins, 19 were indicated as targets for 92 FDA approved drugs. A ranking algorithm was developed based on their level of change of protein expression and availability of literature evidence. This algorithm identified Gemtuzumab, Ozogamicin, Pyridostigmine, Endrophonium, Verapamil, Reteplase, Streptokinase, Tranexamic Acid, Pergolide, Ropinirole, Apomorphine, Rotigotine etc., as potential anti-AD drugs.
Humanising Machines: A beacon of hope for DR market
Artificial Intelligence utilises techniques such as machine learning and deep learning to simulate human characteristics to address complex problems encountered in the computational world. Incorporation of AI in DR has proven to be revolutionary. Machine learning techniques have eased the process of subgroup classification of diagnosis, determination of drug efficacy, ADME prediction, disease target discovery, and decision making, which are the backbone in the various omics approaches. Further, deep learning techniques incorporate artificial neural networks to mine databases using algorithms that aid in unveiling novel therapeutic possibilities. This unique amalgamation of AI with DR, though convenient, is but a start of the transition towards a modern era that involves the integration of the various databases and approaches in DR to minimise clinical trial failures, and provide a safe, economical and effective means to retrieve novel polypharmacological agents.










