The challenge to ensure early access to new drugs and their safe launch is growing globally, particularly after a number of large scale Adverse Drug Reactions (ADRs) were identified and attracted much attention from public, and blockbusters were withdrawn from the market. US FDA intensified its authority of managing post-market safety of new drugs but this resulted in prolonged reviews causing a lot of distress to sponsors. Under such an environment, introducing the unique systems of conditional authorisation and Early Post-marketing Phase Vigilance (EPPV) for new drugs followed in Japan is worth discussing. A mechanism similar to conditional authorisation has recently been introduced in EU, and also in US with the enforcement of FDAAA (FDA Amendment Acts of 2007).
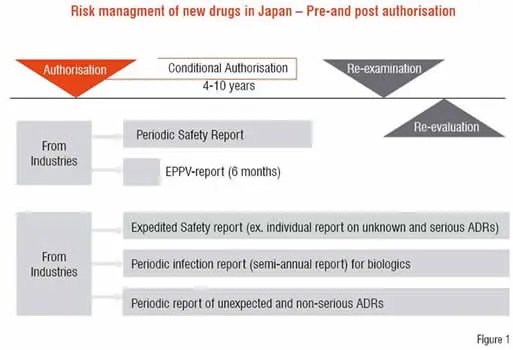
Figure 1 shows the risk management system of new drugs from the authorisation to post-marketing phase in Japan. Certain conditions have been prescribed for authorisation of new drug mainly to ensure its safety, according to its needs. Further to prevent serious ADRs from occurring just after its marketing, it is mandatory to conduct EPPV for six months after the marketing. Other requirements include Expedited Safety Report to collect information such as unknown and serious ADRs, periodic report on infection that was caused by biologics and periodic report of unexpected but non-serious ADRs.
Conditional authorisation
The legal background of conditional authorisation involves Articles 79 and 74-2. The Article 79 was included in the original Japanese Pharmaceutical Affairs Law (PAL) enacted in 1960. The outline is that certain conditions may be prescribed for the authorisation but they shall be confined to the minimum that is required to prevent the occurrence of hazards to public health. Importantly, it says that conditions shall not impose unreasonable obligations on the person intending to obtain it.
The Article 74-2 was introduced by the amendment of PAL in 2002 in which the regulation on conditional authorisation has been strengthened. The background of this revision is that though conditions were appended at the time of authorisation, companies did not necessarily abide by them as there were no punitive provisions for noncompliance. The major revised point is the introduction of a regulation that allows the Ministry of Health, Labour and Welfare (MHLW) to order the revocation or partial change of the authorisation when companies do not comply with the conditions.
Type of the conditions
The first type is the All Cases Surveillance. There are several purposes in conducting this survey. First, this is to obtain high quality information on all the cases to ensure, in particular, safety of the drug. Secondly, it is conducted to detect rare ADRs, which cannot be detected during the drug development phase, by studying a larger number of patients. The Surveillance is also conducted to study the impact the drug may have on the special populations, such as children, patients with hepatic or renal dysfunctions, or elderly people. Furthermore, since information obtained during the drug development phase is from a very limited group of patients reflecting limited circumstances, the information from medical institutions where the drug is administered to patients with various backgrounds becomes extremely important. That is why post-marketing information is necessary and important.
The second type is the condition to conduct additional clinical studies after the drug has been authorised: the practice of post-marketing clinical studies. It may be important to make clear even the frequency of rare ADRs on all drugs in the development phase, or to pursue the possibility of expansion of efficacy claims to include children. However, this kind of work would cause a huge delay in the development. By carrying out these analyses as much as possible in the post-marketing stage, the pre-marketing development time can be made shorter. But in order for that to happen, a strict post-marketing safety control must be in place after the authorisation.
Third one is the access control of the product, i.e. access is limited to specified medical institutions or doctors only, for a specified period of time after the authorisation. The access control on medical institutions also helps prevent the range of the drug use from expanding rapidly before the company has provided adequate information on its product to those institutions.
EPPV
EPPV was introduced in 2001 as a legal requirement (MHLW Ordinance) and now constitutes one type of post-marketing conditions. The conduct of EPPV has been applied, not to an individual product, but uniformly to all new drugs as an obligation.
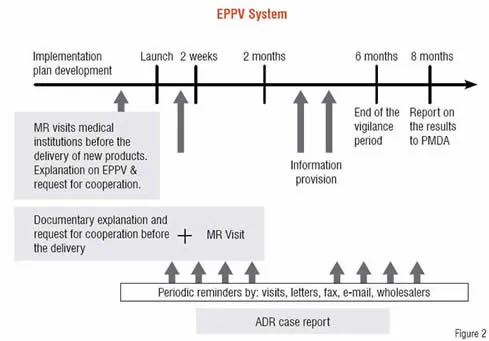
Figure 2 outlines the EPPV system. It is known globally that new drugs have the highest risk at the early post-marketing stage. Before new drug products are delivered to medical institutions, companies provide product information obtained during the development phase to such institutions and request the proper use of the new products. In the event of serious ADRs, the companies request that medical institutions expeditiously report the information to the company. These requests to medical institutions are carried out repeatedly for over six months. This process is intended to prevent medical institutions from starting to use the new products without understanding them, and also to help formulate risk management measures to address possible serious ADRs by speedily collecting information on them.
Though EPPV was introduced in 2001, many patients died from serious ADRs in 2002 because of improper use of the anti-cancer drug, Iressa, gefitinib, just after its marketing. Therefore, MHLW is now working to ensure that the EPPV is thoroughly implemented and it becomes more effective. The review includes the scope of the new drugs to be subject to EPPV, possible extension of the EPPV period that has been six months in principle depending on the type of new drugs, exemption of drugs with expanded indications other than new active substances.
Impacts of the conditions
One of the effects that can be expected from conditional authorisations is increased safety of new drugs. With such conditions, there is a possibility of detecting safety issues including those in long term administration which are not easy to find in clinical studies in the development phase. They may also allow us to check the safety of a new drug in special populations and look into its safety with concomitant medications.
The conditions may lead to the improvement of efficacy. In the development phase, efficacy is often evaluated on the basis of surrogate endpoint(s). However, by doing post-marketing analysis on long-term administration data, efficacy on the true-endpoint, such as the effectiveness on prolonged survival, could be confirmed. Verifying the efficacy of drug for special populations, such as children, may also be possible. In obtaining indication for children, it is more desirable to collect and submit children’s data after marketing, as opposed to delaying the authorisation until all data on children are collected.
Another potential effect of conditional authorisations, i.e. the shortening of the development time is perhaps the most enticing of all for companies. Making wise use of authorisation conditions can lead to shorter development time. The decision on which part of the studies can be conducted in the post-marketing phase, and which part must be done in the development phase, is extremely important both for companies and the regulatory authorities for medicinal products. These will, of course, require case-by-case decisions.
Other effects can also be expected from conditional authorisations. Medical institutions that are allowed to use new drugs under authorisation conditions can prevent serious ADRs caused by their improper use. Eventually, these conditions lead to longer life of the new drugs that were developed with huge investment.
Current status of conditional authorisations
As clinical data of orphan drugs in the development phase are limited, the conditions associated with the authorisations of orphan drugs have been to follow-up all the patients dosed with the drug for a limited or a 10-year period—the drug re-examination period.

For other new drugs, conditional authorisations are given on a case-by-case basis. The authors see the increase of new drug authorisations with conditions prescribed. When conditions are appended, it is obligatory to indicate them on the package insert until the regulatory authority confirms that the obligations under the conditions are met.
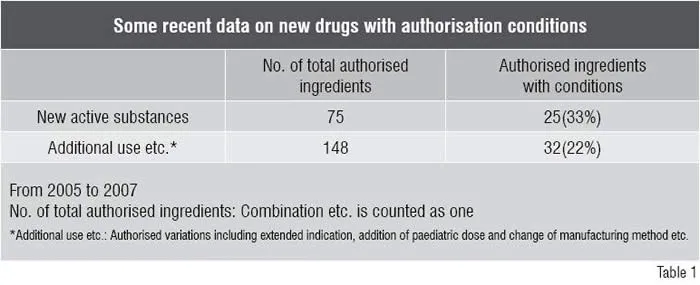
Table 1 shows the recent conditions appended to authorisations in the past three years, from 2005 to 2007. Seventy five new active substances were authorised during the three years, of which 25 (33 per cent of the total) were appended with conditions.
As for other new drugs such as extended indications, 148 ingredients were authorised in the three years and of which 32 (22 per cent of the total) were appended with conditions.
Table 2 shows the type of the requirements in the authorisation conditions in the past three years. Conditions were appended to 57 drugs in total. The conditions include 10 drugs with those of conduct of post-marketing clinical trials such as trials focussing on true endpoint of prolonged survival. Conditions of conduct of post-marketing surveillance were appended to 48 drugs and what were required included “All case surveillance” and “Post-marketing study” aimed at special populations. Access control related to doctors and medical institutions was appended to four drugs and, for 10 drugs, provision of particularly emphasised information on proper use and obtaining informed consent from the patients concerned were required. The other conditions applied to nine drugs include report to the authority of test results conducted in other countries.
Examples of the conditions
An example of Access Control of Medical Institutions and Doctors is Erlotinib, its product name Tarceva. The Condition says “The MAH (marketing authorisation holder) should take appropriate measures to ensure that the drug is only used under the supervision of a physician, a medical centre, all with expertise in lung cancer diagnosis and chemotherapy as well as the potential risks of the drug.”
Interferon Beta-1a, product name AVONEX is an example of Conduct of Post-Marketing Clinical Trial. The Condition says “The MAH should conduct (a) clinical trial(s) to investigate efficacy and safety in long term administration, as a measure of recurrence rate of Multiple Sclerosis, and to report the result to the authority.”
An example of Conduct of Post-Marketing Surveillance on the new drug is Pemetrexed Sodium Hydrate, with product name ALIMTA. The Condition says “The MAH should collect data on safety and efficacy of the drug as well as grasp the background information of the patients by conducting drug use investigation on all the cases until data on a certain number of patients is accumulated after its marketing, because the number of cases in clinical trials in Japan was very limited.”
Future expectations and possibilities of the conditional authorisations
By improving the quality of post-marketing surveillance, the increase in the number of trial subjects needed in the development phase could be curtailed. The authors also think that conditional authorisations could pave the way for a wider use of post-marketing surveillance and test results, so that additional indications could be offered to special populations, such as children.
Another major issue–factors such as ethnicity difference that could be demonstrated by post-marketing surveillance or post-marketing clinical tests should have their information collected in the post-marketing phase as much as possible. It is essential to prevent the extension of development time and the delays in submitting authorisation applications, i.e. “drug lag”, as much as possible.
The wise use of the authorisation conditions as well as EPPV provide a firm basis for better risk and lifecycle management of new drugs.

New Drugs in Japan
Conditional authorisation
Shigeki Tsuda, Executive Director Society of Japanese Pharmacopoeia, Japan
Osamu Doi, Chief Executive
Conditional authorisation and early post-marketing phase vigilance systems constitute an integral part of new drug approval and risk management in Japan. They intend to ensure safe launch and early access to patients of new drugs.
Author Bio

Shigeki Tsuda worked in the Ministry of Health and Welfare, mainly in international affairs and, drug and chemical safety.

Osamu Doi was trained as a biochemist and after working as a researcher in NIH of Japan and in US, he joined the Ministry of Health and Welfare (MHW), where he was one of the founding members of ICH, and left there as the Councilor for MHW.




