
Despite progress on several preventive and therapeutic fronts, cancer remains one of the major causes of morbidity and mortality worldwide. According to the 2011 estimates, about 1.6 million new cases of cancer will be diagnosed and approximately 570,000 deaths will occur due to cancer in the United States (Cancer, Facts and Figures 2011, The American Cancer Society). Surgical resection of the primary tumour, conventional cytotoxic and newer antibody-based chemotherapy, and radiation remain the most widely used clinical strategies for cancer treatment. These approaches, however, have several limitations, including inability of surgical resection to affect distal metastatic disease, toxicity to healthy tissues due to non-specific distribution of chemotherapy and development of multi-drug resistance, and lack of effectiveness of radiation therapy in more aggressive tumours.
Cancer immunotherapy, including the development of prophylactic and therapeutic vaccines, is considered a novel approach for prevention and treatment of cancer. Immunotherapy involves a wide range of options that can ultimately stimulate the body’s immune system to target and eradicate neoplastic cells. The exquisite specificity of the immune system can be utilised to precisely target the cancer cells without affecting normal cells. This hope has motivated researchers over the last several years to develop tumour-specific, prophylactic and therapeutic vaccines, by injecting patients with tumour cells and tumour extracts. Although earlier efforts have met with great disappointments, the rapid increase in knowledge of the immune system and its regulation has led to a resurgence of interest in cancer vaccines and has resulted in several approved products.
The ideal goal of cancer vaccine is to elicit potent anti-tumour immune responses without any contributing side-effects. The major aim is to stimulate both innate and adaptive immunity that can recognise and subsequently eliminate the tumours mass. Cancer immunotherapy continues to be a field of intense research, especially the induction of active immunity. The existence of tumour-specific antigens (TSAs) that can be the targets for an immune response is well established [3]. Advances in cancer genomics anticipate an even greater array of potential TSA targets. To date, a variety of immunization modalities have been employed, including whole tumour cells, tumour lysates, specific tumour antigens, tumour peptides, heat shock proteins, DNA vaccines, exosomes, and dendritic cells (DCs)-based vaccines. As a result, several tumour vaccines showing encouraging results are in the clinics.
In June 2006 the United States Food and Drug Administration (FDA) approved viral oncoprotein-based immunotherapy, known as Gardasil® (or Silgard by Merck). Gardasil® is indicated for the prevention of certain types of human papilloma virus (HPV) infection that is responsible for most HPV-induced anal, vulvar, vaginal, and penile cancers. Gardasil® does not treat existing infection and, therefore, it is used primarily as prophylactic vaccination before adolescence and potential sexual activity. In April, 2010, the FDA approved a DCs-based adaptive immunotherapy, commercially known as Provenge® (or Sipuleucel-T by Denderon Corp.) for the treatment of advanced prostate cancer in men. Provenge® is the first FDA-approved cancer immunotherapy that is targeted towards tumour antigens that exist in the host; therefore, it is used as therapeutic cancer vaccine. Approval of these cancer vaccines has provided the evidence of increased attention to anti-tumour immunotherapy development and, hopefully will lead to additional future successes.
Anti-tumour Immunity
It is well established that the immune system has capacity to attack malignant cells. During malignant transformation cells acquire numerous molecular and biochemical changes converting them vulnerable to immune cells. Yet, it is self-evident that a growing tumour has managed to evade the host defense mechanisms. The exact ways in which the immune system interacts with tumourtumour cells and how cancers are able to escape immunological eradication have only recently started to be fully explained. It is crucial to understand relationship between the tumour and the anti-tumour immune response and how that can be altered to develop successful immunotherapy for patients with cancer. Although anti-cancer immunity involves both the innate and adaptive immune systems, it is generally recognized that CD8+ cytotoxic T lymphocytes (CTL) are the most potent anti-tumour effector cells. The T-cell immune response can be broken down into the following steps. All of the steps need to be satisfied for effective anti-tumour immunity: (1) tumour antigen(s) must be present, and (2) they must be seen as dangerous by the immune system; (3) antigens must be acquired and presented by antigen presenting cells (APC) in the draining lymph node; (4) specific T-cells must then recognize and respond to tumour antigen by proliferation, enter into systemic circulation and reach to the tumour site as CTL; (5) where they need to overcome the local immunosuppressive environment before killing tumour cells. In addition, the memory cells may need to be generated to produce a long-lasting response. It is clear that a growing tumour has managed to escape this process. Failure of the anti-tumour immune response can occur at one or more of these steps. Targeting rate limiting steps with therapies designed to boost the immune response can improve anti-tumour immunity.
Tumour Antigens
Tumours typically express two types of antigen: neoantigens and self-antigens. Neo-antigens (tumour-specific antigens) are derived from mutated self-proteins that are not expressed in normal tissues. Malignant cells express numerous neo-antigens as a result of genomic instability. Most of these mutations do not have functional significance for the tumour cell, but may still provide potential antigenic targets for immune cells. In addition, tumours can also express normal self proteins, but in abnormal quantities or locations (tumour-associated antigens). During T-cell development, T-cell precursors with a strongly self-reactive T-cell receptor are deleted in the thymus, resulting in a T-cell repertoire with a high affinity for foreign antigens and a weak affinity for self antigens. Thus, tumour neoantigens being foreign induce strong immune response whereas tumour associated antigens are considered self and therefore induce weak immune response. CTL responses can be generated to weaker antigens, but require higher antigen concentrations and prolonged duration of exposure.
Role of Antigen Presenting Cells
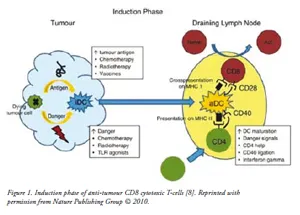
Although T-cell receptors bind with variable affinity to self- and non-self antigens, they are not competent to discriminate dangerous from harmless antigen by themselves. The antigen presenting cells (APCs), especially DCs, satisfy this crucial role by acquiring antigens and responding to associated danger signals. Subsequently, they present the antigens to naïve T-cells in the context of major histocompatibility molecules (MHC) with the appropriate information about the level of danger present as shown in Figure 1. The ‘professional’ APCs present the processed antigen bound to MHC class I molecules to naïve CD8+ T-cells and provide the additional co-stimulation needed to activate CTLs. In addition, helper (CD4+) T-cells recognises the antigen presented on MHC class II and allows DCs to release co-stimulatory signal to promote CTL activation. Thus, helper T-cells offer a ‘second opinion’ to the DCs so that antigens recognized as dangerous are promoted as immunogenic. The fate of the T-cells whether it becomes primed or inactivated as a result of encounter with DCs critically dependent on the state of DCs maturation. Immature DCs are inefficient at cross-presenting antigen and do not express the co-stimulatory molecules required for T-cells activation. DCs maturation is initiated by ‘danger signals’ from antigen and by inflammatory cytokines such as IFN-? [7]. DCs maturation results in increased antigen uptake, up regulation of MHC expression, and expression of co-stimulators CD80 (B7-1) and CD86 (B7-2). During maturation, DCs migrate from tissues to draining lymph nodes where they activate naïve T-cells.
Activation of Anti-tumour Cytotoxic T Lymphocytes
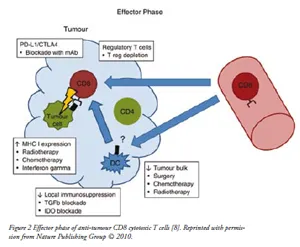
Activation of anti-tumour CD8+ T-cells or cytotoxic T-lymphocytes (CTLs) by mature DCs in the draining lymph node requires several signals: T-cell receptor binding to antigen coupled to MHC class 1, ligation of CD28 on the T-cells with CD80 or CD86 on the DCs and release of inflammatory cytokines such as IL-12 and type 1 INFs. In addition, an effective anti-tumour immunity also requires that CTLs proliferate, survive in the circulation, and enter into the tumour site to execute their effector function as shown in Figure 2. Combining tumour antigen-specific CD4+ T-cells with CD8+ T-cells in adoptive transfer treatment in mice increased the accumulation of tumour-specific CTLs in tumour and lymphoid tissues compared to CD8+ T-cells transfer alone. Thus, it indicated that CD4+ T-cells plays a critical role in anti-tumour CTLs activation process [10]. Persistent CD4+ help and IL-2 secretion are required to maintain CD8+ T-cells function and numbers. In addition, direct cell–cell contact from CD4+ T-cells also protects the effector CD8+ T-cells from activation induced cell death. Moreover, CD4+ T-cell help during CD8+ T-cells priming induces memory CTLs that under go clonal expansion upon re-stimulation with tumour antigen.
Challenges in Cancer Immunotherapy
Tumours are already engaging with the immune system in the patients with cancer. The goal of immunotherapy is to boost the immune response such that the balance shifts from tolerance to rejection. An immunotherapy may fail due to limiting factors at any point in the induction or effector phase. There may be inadequate quantity of tumour antigen present, or vaccination targeting shared, self-antigens may produce only weak T-cells responses that are insufficient to cause tumour regression. In addition, there may be inadequate danger signals to generate strong responses to tumour antigens. The recent clinical trial involving human papilloma virus vaccine showed that anti-tumour immunity mediated through a robust effector T-cells response is essential for successful immunotherapy. In this study the measured T-cells responses strongly correlated with regression of tumour lesions. However, in other clinical trials evaluating tumour vaccines against larger, invasive malignancies the effective generation of tumour antigen-specific CTLs in peripheral blood has not predicted clinical efficacy. This difference may reflect the weaker activity of T-cells generated by cancer vaccines targeting shared self-tumour antigens compared to those directed against viral neoantigens. It may also reflect the presence of a number of barriers to effective immunotherapy in established invasive tumours compared to premalignant lesions. The anti-tumour T-cells response may fail downstream of the induction phase because of: (1) CTLs may remain in the periphery or in the draining lymph node without actually infiltrating the tumour site, (2) CTLs may disseminate to the tumour but are unable to mediate anti-tumour activity, (3) Activated T-cells may fail to continue expansion and maintain effector function, (4) Activated T-cells may be switched off by immune suppressors secreted by tumours, (5) Immune suppressive action of Regulatory T-cells on activated T-cells, (6) tumours may alter their microenvironment to escape immune surveillance.
| Phases of Anti-tumour Immunity |
| Induction Phase: Immature dendritic cells (DCs) acquire tumour antigen migrate to the draining lymph node. Antigen is processed by the DCs and presented to CD4+ T-cells on MHC class II molecules and cross-presented to CD8+ T-cells on MHC class I molecules. DCs activation is promoted by danger signals, IFN- and ligation of CD40 by helper T cells. On activation DCs express co-stimulatory molecules and cytokines, leading to activation of tumour antigen-specific T-cells. |
| Effector Phase: Cytotoxic CD8+ T-cells (CTLs) exit the circulation and enter in to tumour site. CD4+ T-cells facilitate tumour infiltration and may promote secondary expansion of CTLs. Following recognition of the cognate antigen presented on the tumour cells surface, CTLs execute the effector cell function that results in tumour cell killing. Local immunosuppressive mechanisms may inhibit the anti-tumour response, including suppression by regulatory T-cells and inhibitory cytokines. |
Combination Therapy for Cancer Treatment
Although multimodality therapy demonstrated a potential to cure early stage cancers, a key challenge for cancer therapy is to improve outcomes in the patients with advanced disease. Recently, the cancer chemotherapy is considered to synergise with immunotherapy although it often destroys the immune cells. Chemotherapy can have the immune stimulatory effects at number of points during immune response. For example, causing lymphopenia it depletes regulatory T-cells as well as tolerised T-cells to tumour antigens. Homeostatic proliferation to restore immune cell numbers occurs following the cyclical chemotherapy. This phenomenon offers a window to skew the regenerating T-cells response back towards active anti-tumour activity. In addition, chemotherapy induced apoptotic tumour cell death increases the quantity of antigen released and augments antigen cross-presentation by mature DCs to stimulate immune response. Chemotherapy can also sensitise tumour cells that cannot be directly lysed by treatment, to subsequent killing by immune cells. This effect is mediated through up regulation of death receptors such as Fas (CD95) or TRAIL receptors (DR5). Both pre-clinical and clinical studies showed that strategy to combine immunotherapy with chemotherapy reported promising results, however large randomized controlled trials are required to establish the effectiveness. Radiotherapy can also stimulate an immune response, as evidenced by the phenomenon known as the abscopal effect, where in non-irradiated distal metastases shrink following local primary radiation. Radiotherapy has increased MHC 1 peptide presentation of tumours expressed neo-antigens that were recognized by CTLs. This strategy enhanced the effectiveness of a subsequent adoptive transfer immunotherapy. Immunotherapy in the context of surgery is also interesting, with intriguing paradoxical results following complete or partial de-bulking in a mouse model. The de-bulking surgery can reduce tumour burden and tumour-associated immune-suppression to a level in which chemoimmunotherapy can be curative. However, continued antigen presence is needed for memory to be established therefore only partial removal generated long-term anti-tumour memory [19]. This implies that patients currently considered unresectable may benefit from de-bulking surgery as part of a multimodality treatment strategy. In addition, where complete resection is achievable patients may benefit from a continued antigenic stimulus such as a vaccine during adjuvant treatment.
Role of Adjuvant and Vaccine Delivery Systems
Newer generation of vaccines, particularly those based on recombinant proteins and DNA, will have a greater safety profile, but will be less immunogenic that attenuated organisms. An immne-adjuvant is an agent that can stimulate the immune system and increase the response to a vaccine, without having any specific antigenic effect of its own. Immuno-stimulatory adjuvants are predominantly derived from pathogens and often represent pathogen-associated molecular patterns, such as lipopolysaccharides and CpG nucleic acid motifs. Adjuvants activate and engage components of the innate immune system to enhance T and B cell responses. Traditionally adjuvants have been used to increase the magnitude of an adaptive immune response to a vaccine. However, recently adjuvants have been employed to guide the immune system to produce the most effective forms of immunity for each specific pathogen; for example T helper 1 (Th1) cell versus T helper 2 (Th2) cell, CD8+ versus CD4+ T cells, and specific antibody isotypes. Adjuvants are used in cancer immunotherapy to: (1) increase the immune response to a weak tumour-antigen; (2) facilitate the use of smaller doses of antigen; and (3) permit immunization with fewer doses of vaccine. Very few vaccine adjuvants have been licensed for use in human. Alum (aluminum salts) has been widely used for more than 70 years and until recently represented the only approved adjuvant in the United States. MF59 and AS03 (squalane oil in water emulsions) are licensed for adjuvanted influenza vaccines in Europe. AS04, a combination adjuvant composed of monophosphoryl lipid A (MPL) adsorbed to alum is approved for hepatitis B virus (HBV) and HPV vaccines in Europe and has been recently licensed in the USA.
Vaccine delivery systems are generally nano- and micro-particulate systems, such as liposomes, oil-in-water emulsions, and polymeric micro-particles that can encapsulate the antigen payloads and deliver these to the target site and possibly APC’s – either systemically or upon mucosal administration. Increasingly, more complex formulations are being developed in which delivery systems are exploited both for the delivery of antigens and also for the delivery of co-administered immunostimulatory adjuvants. The rationale for this approach is to ensure that both antigen and adjuvant are delivered into the same population of APCs. In addition, delivery systems can focus the effect of the adjuvants onto the key cells of the immune system and limit their systemic distribution, to minimize the potential to induce any adverse effects. The formulation and delivery of potent adjuvants in microparticles may allow the development of prophylactic and therapeutic vaccines against cancers that are currently poorly controlled. In addition, microparticle formulations may also allow vaccines to be delivered mucosally.
Future Outlook for Cancer Vaccines
Tumour cell death can be immunogenic and that can be harnessed to improve cancer treatment outcomes. It may be possible to defeat the issues related to tumour antigen specificity by tailoring immunotherapies to individuals based on tumour gene expression profiles and human leukocyte antigen typing. However, this approach is likely to be both costly and time consuming. A more readily adaptable strategy may be to manipulate the way tumour cells are killed and are sensed by immune cells such that tumour antigens are able to provoke tumour-specific cytotoxic T-cell responses. One can envision a situation in which chemotherapeutic agents are selected to kill tumour cells in a way that is immunogenic, or sensitize tumour cells to immune-mediated cell death. Further strategies may involve deletion of tumour induced immune-suppression to promote the anti-tumour immune response. This might involve tumour vaccines, local inflammatory stimuli or regulatory T-cell depletion. In patients with high tumour burdens, de-bulking surgery may be helpful to reduce the tumour load to a level in which cancer immunotherapy is more effective. Immunotherapies have the potential to become effective treatments for patients with cancer. It seems likely that immunotherapy will be the most efficacious as part of multimodality treatment that may involve chemotherapy, surgery or radiotherapy. Elucidation of the precise mechanisms by which tumour cell death and cancer therapies interact with the host immune response will offer additional knowledge to develop more effective treatments for malignant diseases in the future.






