
The skeleton is one of the most common target organs of metastases in human cancer. Depending on the site of origin of the primary cancer, bone metastases are diagnosed in 23 to 84 per cent of the patients. Although breast, prostate and lung cancers most frequently metastasise to the bone or bone marrow, up to 45 per cent of melanoma patients also develop skeletal metastases. Bone metastases often result in complications such as severe pain, hypercalcemia, pathologic fractures and spinal cord or nerve root compression. Since there is no curative treatment for metastases currently, only palliative treatments such as bisphosphonates, chemotherapy, local radiation, surgery and analgesics are given.
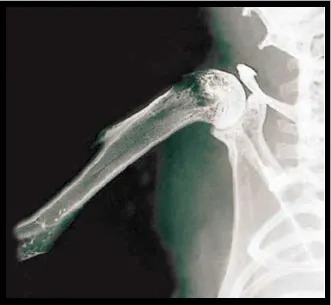
Adoptive Immunotherapy (AIT) may represent an innovative treatment strategy for bone metastases but, thus far, has not been assessed in this setting. The adoptive transfer of in vitro activated and expanded T-lymphocytes has, however, demonstrated significant anti-tumour activity in various animal models using different murine cancer cell lines. Recently, Dudley et al. though not studying bone metastases, also reported promising findings in the clinical setting using adoptive transfer of selected tumour-reactive T-cells in patients with metastatic melanoma following a nonmyeloablative chemotherapy regimen. In the authors' laboratory, a pre-clinical model of bone metastases was evaluated for the study of AIT. This model was modified from Arguello et al. and leads to a high incidence of bone metastases following injection of tumour cells into left cardiac ventricle.
Several properties of the bone contribute to its uniqueness as a metastatic target. Bones constitute a well-vascularised and physically defined microcompartment. Due to the lack of a basement membrane and the presence of fenestrae between the endothelial cells, bone metastases form most frequently within the red bone marrow. In addition, spreading cancer cells might also profit from a rich supply of bone-derived and hematopoietic growth factors. At the same time, the bone marrow is also an important part of systemic immune responses including antigen presentation and various reports describe it as a rich source of protective immune cells in preclinical models investigating active-specific immunotherapy.
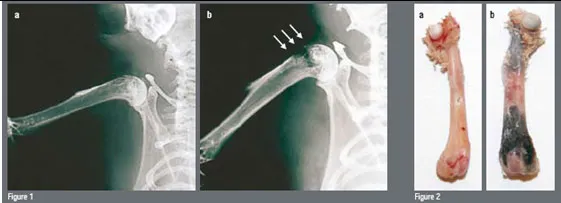
Figure 1: Femur bones (long bones) before and after the skeletal metastase development
In summary, bone and bone marrow with their unique microenvironment support the establishment of tumour micrometastases, but, paradoxically, are also a yielding source of protective immune cells. In patients, the detection of bone marrow micrometastases has been described as an independent predictor for subsequent clinical relapse in distant organs. Several other clinical observations suggest that the bone marrow may be an important site of persistence of dormant tumour cells (or even so-called cancer stem cells) in patients treated with curative intent. Immunologic control of these dormant tumour cells may fail at some point and new metastases may arise. Potential explanations for this phenomenon include impaired immunosurveillance or the development of tolerance.
The data set available on trafficking and function of adoptively transferred T-cells in the bone microenvironment is currently very limited. Several authors have, however, reported on adoptively transferred Tumour Infiltrating Lymphocytes (TIL) and their potential to migrate to metastases throughout the body including the skeleton.
Experimental model of bone metastases
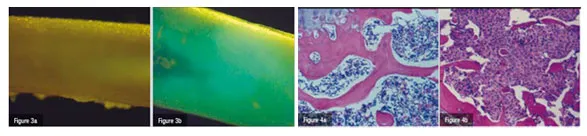
In order to closely mimic the clinical setting, an experimental model of bone metastases was used where tumour cells (breast cancer, lung carcinoma and melanoma) are inoculated into the left cardiac ventricle of immunocompetent laboratory animals under general anaesthesia (Systemic Intra-arterial Administration, SIA). Skeletal metastases develop over time (Figures 1). Tumour colonies of pigmented melanoma cells to the bone can readily be seen on various parts of the skeleton but particularly on well-vascularised portions of long bones, such as the femur (Figure 2b). In a different set of experiments, the authors used melanoma cells that had been transduced with a retrovirus containing the cDNA encoding Green Fluorescent Protein (GFP). This allows for assessment of tumour burden in various regions of the body either by Fluorescence Activated Cell Sorting (FACS) or even in vivo by direct visualisation through fluorescence microscopy (Figure 3).
Generation of effector T-lymphocytes and adoptive immunotherapy
To generate specific effector T-lymphocytes (TE), melanoma cells, that secrete granulocyte-macrophage-colony-stimulating factor (GM-CSF), are injected subcutaneously into the flanks of B6 mice. Eight days following vaccination, the draining superficial axillary and inguinal lymph nodes are harvested. These Tumour Vaccine-Draining Lymph Node cells (TVDLN) are resuspended and activated with an anti-CD3 antibody. After two days of activation the T-cells are harvested and expanded in medium containing IL-2 to generate TE. TE are then harvested, washed and injected intravenously or via SIA into mice with established bone metastases from SIA of D5 melanoma cells.
Regression of bone metastases requires high numbers of adoptively transferred effector T-cells
The adoptive transfer (intravenous infusion) of in vitro activated and expanded T-cells mediated tumour regression in the pre-clinical model of melanoma skeletal metastases used in this study (Figure 2) in a dose-dependent manner. Whereas 10 ? 106 and 50 ? 106 effector T-cells were not sufficient to induce a significant anti-tumour effect in the long bones of mice bearing bone metastases, the adoptive transfer of 100 ? 106 TE cells resulted in macroscopic and microscopic tumour-free distal femurs and proximal tibiae of examined mice (Figure 2a/3/4b). It required a large number of TE cells to induce an anti-tumour effect. It takes half as many TE cells to obtain significant tumour regression in a standard model of pulmonary metastases from the same melanoma cell line as used in the present study.
Furthermore, to observe reproducible metastatic colonisation of the skeleton, injection of 20 times fewer tumour cells into the left heart ventricle is sufficient, compared to intravenous application of 2 ? 105 cells, that is required in the pulmonary metastases model. Low-dose interleukin-2, a cytokine co-administered to support persistence of transferred T-cells, did not influence T-cell mediated regression of bony metastases in the authors' model.
In vivo depletion of CD8+ T-cells abrogates the therapeutic efficacy of AIT for bone metastases
CD8+ T-cells are generally considered to be the main effector population within a specific cellular immune response. In a second set of experiments, CD4+ and CD8+ subsets of T-lymphocytes, were depleted in vivo. Anti-CD4 and anti-CD8 mAB were administered intraperitoneally to deplete the respective T-cell population. Because the depleting antibodies were given after the adoptive transfer,
CD4+ and CD8+ subsets of both the transferred and endogenous population of cells were depleted by at least 97 per cent as measured by flow cytometry. Bone metastases were detectable in only one animal in the IgG antibody control group and one animal in the group receiving the antibody for the depletion of CD4+ cells. In contrast, all animals receiving in vivo depletion of CD8+ cells exhibited pigmented tumour colonies on long bones. The histological examination of long bones, which was performed 17 days following SIA of tumour cells showed the bone marrow cavity to be almost entirely occupied by tumour cells (Figure 4b). Without CD8+ depletion, tumour cells were undetectable and the bone marrow appeared normal (Figure 4a), clearly demonstrating the key role of CD8+ cells in the immune-mediated regression of bone metastases in this setting.
Efficiency depends on the route of administration
The authors hypothesised that intravenous injection might limit trafficking of TE to bone or bone marrow due to early entrapment in the lungs. To circumvent this problem, TE cells were also administered directly into the arterial systemic circulation via the left cardiac ventricle bypassing the pulmonary microvasculature. 50 × 106 TE cells failed to mediate regression of bone metastases when injected intravenously. In contrast, transfer of the same number of TE cells into the arterial circulation through the left cardiac ventricle completely cleared bones of visible tumour colonies. But using even lower number of TE cells (25 × 106) did not lead to a significant reduction in animals with bone metastases.
TE traffic to bone or bone marrow following IV injection and division of transferred cells is detectable
Since AIT had proven to be an effective treatment protocol for experimental metastases to the skeleton in this model, it was hypothesised that this effect was mediated by the transferred TE and that these cells were able to migrate to the bone marrow of animals bearing 3-day bone metastases. To address this question, TE cells were generated from B6. PL-Thy1a/Cy mice, which carry the T-lymphocyte-specific Thy1a (Thy1.1) (CD90.1) allele. Thus, donor T-cells could be easily distinguished from recipient T-cells using flow cytometric analysis.
To assess cell division of transferred cells, TE cells were labelled with CFSE before adoptive transfer. Not surprisingly, in tumour-bearing animals, adoptively transferred cells were present in the spleen after 24 hours following IV injection. Sixteen per cent of all CD8+ cells originated from Thy1.1 donor mice at this point of time. This number declined to 8 per cent by day 3 and 3 per cent by day 6 following AIT. A sequential reduction of CFSE fluorescence was detected in the spleen at all time points indicating that transferred cells had also divided. Thy1.1+ / CD8+ donor cells were also present in the bone marrow at each of the three times of measurement with 18 per cent of CD8+ cells in the bone marrow deriving from the transferred population after 24 hours. This number was reduced to and remained at 5 per cent on day 3 and 6. At two of the three time points, CFSE fluorescence indicated that Thy1.1+ had undergone cell division in the bone marrow. Since divided donor cells were also detectable in peripheral blood of animals on days 3 and 6, it remained uncertain if cell division had occurred within the bone or bone marrow or if donor cells had migrated to this location after dividing elsewhere.
Summary and perspective
Thus far, the adoptive transfer of TE to animals with systemic metastases has demonstrated therapeutic efficacy in disease models as different as melanoma, colorectal and brain cancer. However, to date, no attempt has been made to define premises for the investigation of adoptive T-cell transfer as immunotherapeutic approach to bone metastases. In this study, the therapeutic value of adoptive immunotherapy in a model of metastatic disease to the skeleton has been demonstrated for the first time. Further investigation seems obvious considering the fact that the skeleton is among the most common sites of metastatic disease and that skeletal metastases represent a devastating complication for patients.
The sheer presence of single tumour cells in the bone marrow has been identified as a (poor) prognostic indicator for the development of distant metastases following curative treatment of the primary tumour. In general, the rationale for adjuvant therapy is based on the assumption that clinically undetectable, viable tumour cells are present at distant sites, e.g. the bone marrow, at the time of curative treatment. One key feature of micrometastatic tumour cells in the bone marrow is, however, their relative resistance to chemotherapeutic agents. In breast cancer patients this was attributed to overexpression of the erbB-2 proto-oncogene possibly inducing tumour cell resistance. In addition, most of the tumour cells isolated from the bone marrow failed to express typical proliferation markers such as Ki-67 and p120 indicating that these cells might be resting in the G0 phase of the cell cycle and therefore be less susceptible to cytotoxic agents. The immunotherapeutic approach used in this study might represent a cell-cycle independent treatment modality which can recognise and eradicate tumour cells in the bone or bone marrow independent of their state of proliferation.
However, as one can imagine, there are major obstacles to translating these strategies into a clinical application. In conclusion, adoptively transferred TE cells showed a remarkable ability to migrate to bone or bone marrow and mediate tumour regression in the authors' pre-clinical model of skeletal metastatic disease. Therefore, further studies of adoptive T-cell immunotherapy for macrometastatic and micrometastatic disease to the skeleton seem warranted with the long-term objective to implement this cell-cycle independent treatment regimen as a valuable addition to established treatment modalities.
Acknowledgements: This work was supported by the Chiles Foundation, Portland, Oregon, USA, NIH grants CA80964, CA92254, CA107243, CA119123 and the Walter-Schulz-Foundation, Munich, Germany.
References
1. Coleman RE. Skeletal complications of malignancy. Cancer 1997; 80: 1588-1594.
2. Tong D, Gillick L, Hendrickson FR. The palliation of symptomatic osseous metastases. Final results of the study by the Radiation Therapy Oncology Group. Cancer 1982; 50: 893-899.
3. Janjan N. Bone metastases: Approaches to management. Semin Oncol 2001; 28 (4 Suppl 11): 28-34.
4. Winter H, Hu H-M, Urba WJ et al. Tumor regression after adoptive transfer of effector T cells is independent of perforin or FAS ligand (APO-1L/CD95L). J Immunol 1999; 163: 4462-4472.
5. Lechaux D, Gervais A, Dazord L et al. Adoptive immunotherapy monitored by micro-MRI in experimental colorectal liver metastasis. Anticancer Res 2002; 22: 151-158.
6. Inoue M, Plautz GE, Shu S. Treatment of intracranial tumors by systemic transfer of superantigen-activated tumor-draining lymph node T cells. Cancer Res 1996; 56: 4702-4708.
7. Dudley ME, Wunderlich JR, Robbins PF et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002; 298: 850-854.
8. R?ttinger D, Li R, Urba WJ et al. Evaluation of a preclinical model of bone metastasis for the study of adoptive immunotherapy. Eur Surg Res 2003; 35: 346-351.
9. Arguello F, Baggs RB, Frantz CN. A murine model of experimental metastasis to bone and bone marrow. Cancer Res 1988; 48: 6876-6881.
10. De Bruyn PPH. Structural substrates of bone marrow function. Semin Hematology 1981; 18: 179-193.
11. Orr FW, Sanchez Sweatman OH, Kostenuik P et al. Tumor-bone interactions in skeletal metastasis. Clin Orthop 1995; 312: 19-33.
12. Doe B, Selby M, Barnett S et al. Induction of cytotoxic T lymphocytes by intramuscular immunization with plasmid DNA is facilitated by bone marrow-derived cells. Proc Natl Acad Sci 1996; 93: 8578-8583.
13. Hornung RL, Longo DL, Bowersox OC et al. Tumor antigen-specific immunization of bone marrow transplantation donors as adoptive therapy against established tumor. J Natl Cancer Inst 1995; 87: 1289-1296.
14. M?ller M, Gounari F, Prifti S et al. EblacZ tumor dormancy in bone marrow and lymph nodes: active control of proliferating tumor cells by CD8+ immune T cells. Cancer Res 1998; 58: 5439-5446.
15. Pantel K, Izbicki J, Passlick, B, et al. Frequency and prognostic significance of isolated tumour cells in bone marrow of patients with non-small-cell lung cancer without overt metastases. Lancet 1996; 347: 649-653.
16. Schlimok G, Funke I, Bock B et al. Epithelial tumor cells in bone marrow of patients with colorectal cancer: immunocytochemical detection, phenotypic characterization, and prognostic significance. J Clin Oncol 1990; 8: 831-837.
17. Jauch KW, Heiss MM, Gruetzner U et al. Prognostic significance of bone marrow micrometastases in patients with gastric cancer. J Clin Oncol 1996; 14: 1810-1817. 18. Dillman RO, Hurwitz SR, Schiltz PM et al. Tumor localization by tumor infiltrating lymphocytes labeled with indium-111 in patients with metastatic renal cell carcinoma, melanoma, and colorectal cancer. Cancer Biother Radiopharm 1997; 12: 65-71.
19. Griffith KD, Read EJ, Carrasquillo JA et al. In vivo distribution of adoptively transferred indium-111-labeled tumor infiltrating lymphocytes and peripheral blood lymphocytes in patients with metastatic melanoma. J Natl Cancer Inst 1989; 81: 1709-1717.
20. Lyons AB. Analysing cell division in vivo and in vitro using flow cytometric measurement of CFSE dye dilution. J Immunol Methods 2000; 243: 147-154.
21. Braun S, Kentenich C, Janni W et al. Lack of effect of adjuvant chemotherapy on the elimination of single dormant tumor cells in bone marrow of high-risk breast cancer patients. J Clin Oncol 2000; 18: 80-86.
22. Braun S, Pantel K, Muller P et al. Cytokeratin-positive cells in the bone marrow and survival of patients with stage I,II, or III breast cancer. N Engl J Med 2000; 342: 525-533.
23. Hancock MC, Langton BC, Chan T et al. A monoclonal antibody against the c-erbB-2 protein enhances the cytotoxicity of cis-diamminedichloroplatinum against human breast and ovarian tumor cell lines. Cancer Res 1991; 51: 4575-80.
24. Pantel K, Schlimok G, Braun S et al. Differential expression of proliferation-associated molecules in individual micrometastatic carcinoma cells. J Natl Cancer Inst 1993; 85: 1419-1424.
25. Zhu H, Melder RJ, Baxter LT et al. Physiologically based kinetic model of effector cell biodistribution in mammals: implications for adoptive immunotherapy. Cancer Res 1996; 56: 3771-3781.








