
The desired features of pharmaceutical drug delivery for intravenous administration (their small size, biodegradability, high content of a drug in a final preparation, prolonged circulation in the blood, and the ability to target required areas) are reasonably well met by liposomes, microcapsules and nanoparticles for water-soluble drugs. The development of nanoparticulate drugs displaying all of these properties for poorly soluble pharmaceuticals still represents a challenge. Low solubility in water, however, tends to be an intrinsic property of many drugs, including some powerful anti-cancer agents. Intravenous administration of relatively large aggregates of an insoluble drug may result in embolisation of these particles into small blood capillaries and may cause unwanted effects like tissue ischemia. Hence, it does not allow for achieving therapeutically significant concentrations.
Many promising drug candidates never enter further development processes because of solubility problems. On multiple occasions, micelles can serve as drug delivery systems for poorly soluble pharmaceuticals. However, there are problems, which include low loading efficacy of the drug into the micelles; problems with controlling the release rate of the drug, and with micelle stability.
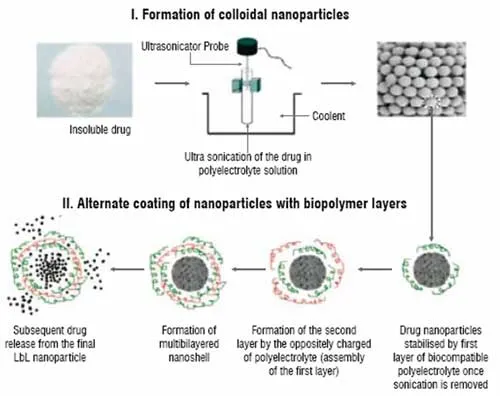
Figure 1: Scheme of LbL nanocolloidal particles formation from insoluble drugs
Optimal drug delivery and reduction of systemic adverse effects have been age-old problems in chemotherapeutics of human cancer. With the advent of the use of water insoluble cancer drugs like paclitaxel and tamoxifen, it has been realised that better formulations are still needed for more specific and controlled delivery of these agents. In a novel approach to form stable nanocolloids of these drugs with high content of the active drug and controllable release rate, using a sonicated Layer-by-Layer (LbL) polyelectrolyte coating technology is suggested. This is development of traditional LbL microencapsulation based on alternate adsorption of oppositely charged components (linear polyelectrolytes, proteins, and nanoparticles). To achieve nanosize cores, aqueous suspensions of poorly soluble drugs are subjected to the powerful ultrasonic treatment. To keep the nanoparticles formed under the sonication from fast re-aggregation, they are stabilised in a solution by sequential addition of polycations and polyanions and by assembling ultra-thin polyelectrolyte shells on them (Figure 1). LbL assembly allows preparation of multilayer shells with thickness of 5 to 50 nm and necessary composition. In this process, a nano-architectural approach for designing shells of different components, including ones serving as diffusion barrier and outermost layers containing targeting agents, was realised.
The procedure using synthetic polyelectrolyte was first established and then was transferred to biocompatible polymers such as cationic (polylysine, protamine sulphate) and anionic (dextran sulfate, and bovine serum albumin). After depositing the first polycation layer on the surface of a drug nanoparticle, an oppositely charged polyanion is added. This results in the formation of a stable inter-polyelectrolyte complex shell around each drug nanoparticle. By varying the charge density of each polymer or the number of coating cycles, particles with a different surface charge and different composition can be prepared. The use of a polymer containing reactive groups (such as amino groups) for the last "outer" surface layer allows for the attachment of specific ligands and other moieties of interest to drug nanoparticles. This article presents a proprietary method (Nemucore Medical Innovations Inc., Boston) of sonicated LbL assembly for the preparation of stable nanocolloids of paclitaxel and tamoxifen with very high drug content.
Materials and methods
Poorly soluble and potent anti-cancer drugs paclitaxel and tamoxifen have been used. Polyelectrolyte that was used included positively charged polylysine, poly(dimethyldiallyl ammonium chloride) (PDDA), protamine sulphate (PS); and negatively charged sodium poly(styrene sulphonate) (PSS) and bovine serum albumin (BSA). Ultra Sonicator 3000 (Misonix Inc) was used for drug crystals disintegration at the power of 18W. To prevent the sample from overheating during the sonication and to keep the temperature in the range of 20-25?C, liquid nitrogen was used to cool the sample tubes.
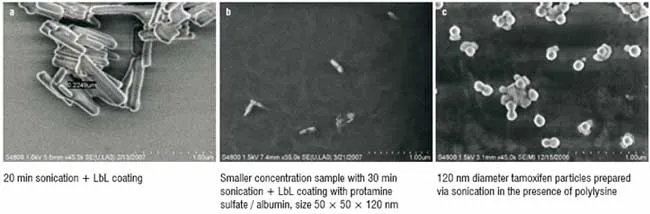
Figure 2: SEM images (Hitachi-2006) of LbL encapsulated drug nanoparticles (Paclitaxel)
For disintegration, all drug samples were ultrasonicated at reduced temperatures for 10 minutes before polyelectrolyte was added. Once this was added, the samples were sonicated for another 20 minutes. Polycations were used to form the first surface layer, since drug nanoparticles were found to bear the intrinsic negative charge. Drug samples were then centrifuged, washed and re-suspended in PBS buffer to get rid of the excessive polycation before further zeta potential readings were taken. Then, the coating process was repeated with polyanion, followed by polycations, and so on. The resultant high-concentration drug colloids were kept in a low volume of saturated solution to prevent drug release. To study the release rate of drugs from the colloidal particles, the samples were placed in horizontal diffusion chambers made of cellulose acetate membrane and stirred in large volume of PBS, pH 7.2, to mimic sink conditions expected in vivo.
Attachment of ligand moieties to the LbL nanocolloids of poorly soluble drugs
To prepare nanocolloids with the "reactive" surface suitable for the covalent attachment of various ligands to their surface, polycation containing free amino groups was used to form the outer layer on drug nanoparticles. To conjugate the monoclonal nucleosome-specific 2C5 antibody (mAb 2C5) recognising a broad variety of cancer cells, the reaction was carried out in two steps. In the first step, the carboxylate groups on the mAb 2C5 were activated using 1-ethyl-3-carbodiimide hydrochloride (EDC) and N-hydroxysulfosuccinimide (sulfo-NHS) to make it amine-reactive. In the second step, the activated antibody was added to LbL paclitaxel nanoparticles coated with polyamino-containing polymer. To verify the preservation of mAb 2C5 specific activity after the conjugation with LbL-paclitaxel nanoparticles, a standard ELISA was performed. The cytotoxicity of various concentrations of LbL-paclitaxel nanoparticles against and MCF-7 and BT-20 cells was studied using a MTT test.
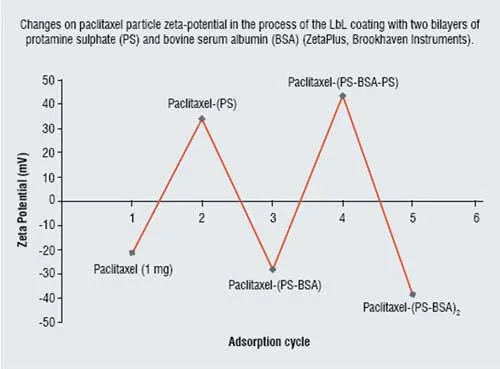
Figure 3: Changes on paclitaxel particle zeta-potential
Results and discussion
After 30 minutes of LbL-assisted sonication, paclitaxel particle sizes of about 50 ? 50 ? 120 nm were obtained. Further increase in the sonication time did not result in significant decrease in drug particle size (Figure 2). The charged bare drug nanoparticles, which were negative initially became positive and formed stable colloids after the sonication with the addition of cationic polylysine or protamine sulphate. Further, assembly with the corresponding changes in zeta potential values allowed formation of the two bilayer shell with biocompatible protamine sulfate and albumin: (PS/BSA)2. Figure 3 gives values of the surface zeta potential during the process of sequential four-step protamine sulfate / albumin adsorption on paclitaxel nanocores. Thickness of such shell estimated with Quartz Crystal Microbalance was ca. 6 nm in dry state and 12 nm in water. Similar Zeta potential alternation was observed during shell assembly on tamoxifen nanoparticles.
Nanoparticle imaging and drug release
The particle size of all the drug samples formulated by the LbL technology into nanocolloidal state was confirmed by scanning electron microscopy and confocal fluorescent microscopy (Figure 2). At 20 min sonication, submicron particles were obtained, and further sonication with polycations and liquid nitrogen cooling resulted in nanosized particles. LbL-coated paclitaxel has the elongated rod-like shape of approximately 50 ? 50 ? 120 nm. Particles of tamoxifen demonstrate mainly a spherical shape and have a diameter of 120 ? 30 nm. Tamoxifen's spherical shape probably is due to melting and then solidifying of paclitaxel nanoparticles during powerful sonication (instantaneous local temperature during collapsing cavitations' microbubbles reaches hundreds of degrees). This was confirmed by the amorphous form of the resultant tamoxifen nanoparticles. The SEM images were obtained after drying the samples, and during this process the nanoparticles become partially aggregated. This aggregation does not proceed in aqueous suspension, as it was demonstrated with fluorescence confocal images of the samples.
Taking into account that the thickness of a single polymeric layer is approximately 3 nm in dry state, we can calculate that the drug content in stable nanocolloidal drug particles is from 90 per cent wt (in case of paclitaxel particles with a double layer coating) to 85 per cent wt (in case of tamoxifen particles with a triple bilayer coating) which is dramatically higher compared to other solubilisation methods. Colloidal suspensions of both drugs were stable during a month of observation.
LbL technology allows control of drug release rate from polymer-stabilised colloidal nanoparticles by simple changes in coating thickness or composition. As one could expect, a slower release rate is observed as the number of polyelectrolyte layers in the shell increases. At sink conditions, non-coated tamoxifen particles (after sonication) dissolve within approximately two hours, while LbL coating allows to extend this time to 10 hours. Figure 4 gives the release curves for 2 mg/mL tamoxifen in PBS buffer at pH 7.2 through a 0.2 micrometer pore nitrocellulose membrane. Two upper curves represent bare uncoated micronised drug, and two lower curves show release from 125-nm diameter nanoparticles coated with one bilayer and PDDA monolayer and thicker shell of three bilayers of (PDD/PSS)3. Similar results were obtained for paclitaxel also. These are initial release experiments done with synthetic polymers. Experiments with biocompatible shells of (protomine sulfate / albumin)3 gave similar release rate.
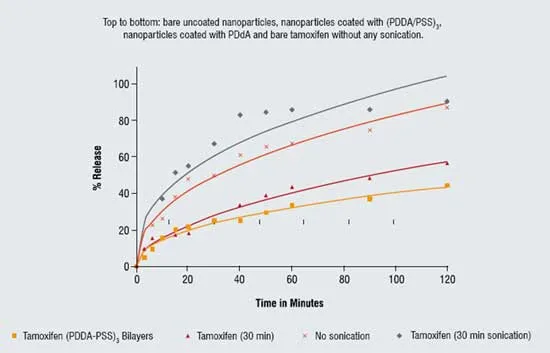
Figure 4: Release of tamoxifen nanocapsules at sink condition
Surface modification of LbL-coated drug nanoparticles
To confirm that LbL-coated drug nanoparticles can be easily derivatised on the surface in order to impart them various additional properties including targeted delivery, tumour-specific mAb 2C5 have been attached to the paclitaxel nanoparticles via free amino groups belonging to the surface layer of PAH. ELISA with the nucleosome monolayer (specific antigen for mAb 2C5), the results of which clearly confirms that 2C5-modified LbL-coated paclitaxel nanoparticles acquire the ability to specifically recognise the target antigen, i.e. become targeted.
Increased cytotoxicity of tumour cell-targeted LbL drug nanocolloids in vitro
In vitro experiments with MCF-7 and BT-20 cancer cell lines clearly confirmed that targeted drug nanoparticles demonstrate higher cytotoxicity than the non-targeted counterparts. At paclitaxel concentration of 100 ng/mL in case of MCF-7 cells and 30 ng/mL in case of BT-20 cells, when virtually no cytotoxic effect can be observed with non-targeted LbL-coated paclitaxel nanoparticles (around 95 per cent of cancer cells remain alive after the incubation for 48 or 72 hours), 2C5-targeted LbL-coated paclitaxel particles kill at least 30 per cent of cancer cells. This result confirms that LbL technology allows for decorating the surface of stable colloidal drug particles with very high drug content with various additional functions as needed.
Results
In conclusion, the following formulation technique was demonstrated: 1. Stable nanocolloids of insoluble drugs with very high drug content can be prepared through the application of the sonicated LbL technology, i.e. combination of ultrasonication and alternate adsorption of oppositely charged polyelectrolytes, resulting in coated nanoparticles with the content of the drug far exceeding other known systems; 2. Drug release rate from such nanoparticles can be controlled by assembling organised multilayer shells with required wall composition, density and thickness; 3. Various additional functions, such as specific targeted ligands, can be attached to the surface on nanocolloidal particles of poorly soluble drugs by using a polymer with free reactive groups for the "outer" coating and preserve their specific properties upon the attachment; 4. Since drugs are not modified in any way in the process of solubilisation and release as free drug molecules, there is no concern regarding any possible change in drug activity in vivo, however, to deliver a desired dose of a poorly soluble drug in the body, a very small quantity of polymeric carrier is required compared to any other protocol currently used for administration of poorly soluble pharmaceuticals.
References
1. V. Torchilin, Multifunctional nanocarriers, Adv. Drug Deliv. Rev. 58 (2006) 1532-1555.
2. A. Domb, Y. Tabata, M. Ravi Kumar, S. Farber (Eds.), Nanoparticles for Pharmaceutical Applications, American Scientific Publ., Stevenson Ranch, CA, 2007.
3. C. Lipinski, F. Lombardo, B. Dominy, P. Feeney, Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings, Adv. Drug Deliv. Rev. 46 (2001) 3-26.
4. V. Torchilin, Micellar Nanocarriers: Pharmaceutical Perspectives, Pharm. Res. 24 (2007) 1-16.
5. E. Donath, G. Sukhorukov, F. Caruso, S. Davis, H. Moehwald, Novel Hollow Polymer Shells by Colloid - Templated Assembly of Polyelectrolytes, Angew. Chem, Int., 37 (1998) 2202-2205.
6. H. Ai, S. Jones, M. DeVilliers, Y. Lvov, Nano-encapsulation of Furosemide Microcrystals for Controlled Drug Release, J. Control. Release 86 (2003) 59-68.
7. A. Agarwal, Y. Lvov, R. Sawant, V. Torchilin, Stable Nanocolloids of Poorly Soluble Drugs with High Drug Content Prepared Using Layer-by-Layer Technology. J. Control. Release 91 (2008) 972-977.
Read the full article — it's free
Register with Pharma Focus Asia to unlock expert insights, research articles, and in-depth industry analysis.








