
Despite billions of dollars spent on research every year, cancer remains one of the leading causes of death worldwide. Understanding of the molecular mechanisms underpinning many cancers has progressed rapidly, but our ability to treat the disease remains limited. Most cancer therapeutics rely upon the ability of the agent to eradicate tumour cells successfully from the patient, but this almost always results in the unnecessary and unwanted death of “normal” bystander cells. Moreover, due to the nature of many of these drugs, the highly proliferating normal cells of the body tend to be the most affected, including those of the bone marrow and the immune system, resulting in an immunocompromised patient who is then highly susceptible to life-threatening pathogens. Thus, it is clear that efficient and above all specific anti-cancer agents are urgently required.
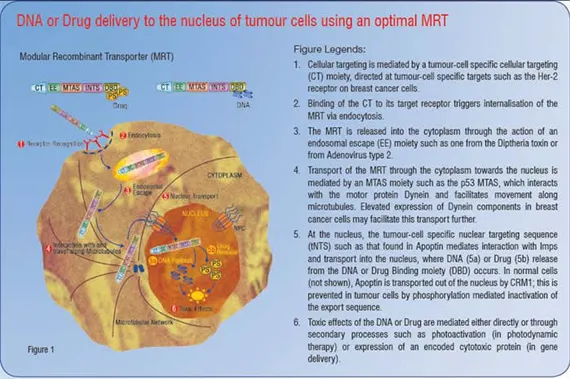
Delivering drugs to the nucleus: Lessons from viruses
An effective means to mediate efficient tumour-cell death will be to deliver drugs specifically to tumour cells, and subsequently to direct them to hypersensitive subcellular compartments within these cells. This will avoid harmful side effects on normal healthy cells without compromising on tumour cell cytotoxicity. In order to achieve this, one needs to overcome a series of natural barriers, which prevent the entry of foreign material into all cells and more importantly into the nucleus within these cells. Viruses have evolved mechanisms to overcome such barriers, many being able to deliver their genomes into the nucleus of host cells. However, safety considerations such as pathogenicity and stimulation of an immune response hamper their use in clinical settings. To overcome these issues, Modular Recombinant Transporters (MRTs) are being developed that mimic viruses by retaining all of the necessary cellular and subcellular targeting functions of viruses, but without the safety concerns.
The first step in drug delivery is cell entry, which involves binding to and passage through the cellular membrane. To achieve this, many viruses utilise recognition of specific receptors expressed on the surface of target cells. Depending on the virus or vector in question, it may be internalised into an endosome subsequent to receptor binding as is the case for non-enveloped DNA viruses such as Adenovirus. Endosomal entrapment will ultimately result in degradation of the enclosed ligand when the endosome fuses with a lysosome.
To overcome this, many viral proteins function as endosomal escape moieties by undergoing conformational changes in response to a drop in pH, and thus facilitating membrane disruption and release of the virus into the cytoplasm. The virus must subsequently traverse the cytoplasm through the crowded network of the cytoskeleton, avoid degradation, and translocate into the nucleus in order to be expressed and replicated, or in some cases, integrated into the host cell genome, as is the case for retroviruses. Nuclear transport is known to be the most rate-limiting step in this process.
The nucleus is the heart or control centre of the eukaryotic cell, being the site of storage and replication of the cell’s genetic material, and of processes such as transcription and ribosome assembly that are central to synthesising the cellular complement of proteins that carry out all of its functions. The nucleus is separated from the rest of the cell cytoplasm by a double membrane known as the Nuclear Envelope (NE), punctated by Nuclear Pore Complexes (NPCs), through which all transport between the nucleus and cytoplasm occurs. Small molecules are able to pass through the NPC via passive diffusion, but molecules > ~45 kDa require specific targeting signals to gain either access to or egress from the nucleus. The signals for transport in the import direction, known as Nuclear Localisation Signals (NLSs), are recognised by members of the importin (Imp) superfamily of nuclear transport receptors. NLSs tend to be similar in nature, usually consisting of one (monopartite) or two (bipartite) short stretches of basic amino acids, typified by the NLS of the Simian Virus 40 Large Tumour Antigen (T-ag: PKKKRKV132) or that of the Xenopus protein nucleoplasmin (KRPAATKKAGQAKKKK170) respectively. Conventional nuclear transport involves recognition of the NLS by the Imp ? subunit of the Imp heterodimer, followed by docking at and translocation through the NPC mediated by the Imp ? subunit. Once inside the nucleus, binding of the monomeric guanine nucleotide binding protein Ran, in its GTP bound form, to Imp ? actively displaces Imp ? and effects release of the cargo into the nucleoplasm. Imp ? itself and its numerous homologues can also mediate nuclear transport of various cargoes in an analogous fashion to Imp ?/?, but without the need for Imp ?. Nuclear export is an analogous process to nuclear import, requiring Nuclear Export Signals (NESs), which are recognised by Imp ? homologues known as exportins, of which exportin-1 (CRM-1) is the best known example.
Effective drug delivery agents need to combine elements of each of the above functions. In tumour-cell specific delivery, they additionally need to possess selectivity for cancer cells. For example, ligands for cell-surface receptors that are specific to or upregulated on tumour cells could be included as Cell Targeting (CT) moieties in an MRT system. When combined with an Endosomal Escape (EE) moiety and an effective NLS, an efficient drug or gene delivery agent can be produced. Examples of such MRTs have already begun to emerge, some of which are capable of delivering reporter genes or drugs specifically to tumour cells in culture, as well as targeting expression of a luciferase reporter plasmid to the liver of mice following an intravenous injection.
NLSs as a key to efficient nuclear delivery
It is becoming increasingly clear that nuclear transport of DNA is the key determinant of effective gene delivery, and that increasing nuclear delivery by incorporating an efficient NLS results in significantly higher levels of overall gene expression in most cases. For example, we have recently developed an efficient non-viral gene delivery method based upon engineered histones, where optimised nuclear targeting of the constructs through inclusion of an optimised T-ag NLS resulted in a significant increase in transgene expression. Similarly, inclusion of the T-ag NLS to an MRT containing ?-galactosidase as a carrier, poly-lysine as a DNA compacting agent and the foot-and-mouth disease virus RGD sequence as a CT moiety resulted in a 30-fold increase in transfection efficiency of CaCo2 cells when compared to the MRT lacking the T-ag NLS.
In the case of drug delivery, the nucleus is also often the most sensitive site for drug induced damage, such as in the case of photosensitising agents used in Photodynamic Therapy (PDT). PDT relies upon the specific activation of photosensitisors such as protoporphoryn IX or chlorin e6 using long wave length light, releasing singlet oxygen species, the cytotoxic effects of which do not usually exceed 40nm from the site of activation.
Since the nucleus is a hypersensitive site for active oxygen species-induced damage, coupling of the optimised T-ag NLS to chlorin e6 containing complexes, either cross-linked to a carrier or encoded as part of a fusion protein can result in a reduction of the EC50 by a factor of 2000-fold, highlighting the importance of specific nuclear delivery of these chemical agents. In terms of cancer therapy, delivery of these active agents specifically to the nucleus of tumour, but not normal cells, is the key to their being “magic bullets” in terms of effective and safe anti-tumour therapeutics.
Tumour cell-specific nuclear targeting
Several proteins have been demonstrated to kill tumour cells selectively, including the Tumour Necrosis Factor Related Apoptosis Inducing Ligand (TRAIL) and the adenovirus type 2 Early region 4 open reading frame 4 (E4orf4). TRAIL specifically kills malignant cells, but spares normal tissues, by binding to death receptors expressed exclusively in the cancer cell membranes and triggering apoptosis through a mitochondria-independent signalling pathway. Nuclear localisation appears to be important for the efficiency of killing, but is not essential for E4orf4 induction of p53-independent apoptosis. Chicken Anaemia Virus Viral Protein 3 (CAV VP3, also known as Apoptin) is intriguing, because it is believed to induce tumour cell-selective cell killing, specifically dependent on its ability to localise specifically in tumour, but not normal cells.
Apoptin is a 121-amino acid protein, which can induce apoptosis in chicken thymocytes and several human tumour but not normal cells, through what appears to be a p53-independent, Bcl-2 resistant pathway. However, the use of non-isogenic cell lines in several studies, combined with reports demonstrating apoptosis in non-transformed primary human fibroblasts suggest that Apoptin-induced apoptosis may not be specific to tumour cells. Nonetheless, Apoptin’s preferential localisation in the nucleus of tumour/transformed cells makes it an intriguing possibility as a targeting moiety for cancer therapy applications.
Apoptin contains a basic bipartite NLS in its C-terminus (residues 80-121), which is necessary and sufficient for nuclear accumulation. Interestingly, when the N-terminus of Apoptin is removed, Apoptin (a.a. 74-121) binds to Imp ? with 3-fold higher affinity, implying that Apoptin’s nuclear accumulation may be regulated by intramolecular masking of the NLS through the N-terminus. Apoptin also posses es a CRM-1 recognised NES (a.a. 97-105) located between the two basic clusters of the bipartite NLS, which appears to be the basis for tumour-cell enhanced nuclear localisation of Apoptin.
Most importantly, the NES is regulated by phosphorylation at residue T108 in tumour cells, but not normal cells, whereby phosphorylation of this residue in tumour cells inhibits NES-mediated nuclear export of Apoptin, resulting in increased nuclear accumulation. Thus, residues 74-121 encompassing both the NLS(s) and the phosphorylation regulated NES represent a tumour cell-specific Nuclear Targeting Signal (tNTS), responsible for Apoptin’s enhanced nuclear localisation in tumour cells. Clearly the tNTS is an exciting prospect in a therapeutic context to deliver drugs or DNA specifically to the nucleus of tumour cells, thereby increasing the safety and efficacy of anti-tumour therapies.
A new class of MRTs: tNTS at the forefront?
The fact that it confers preferential accumulation in the nucleus of tumour as opposed to normal cells makes the Apoptin tNTS a very attractive possibility for use in a modular therapeutic system. However, Apoptin can accumulate in the nucleus of normal cells, but to a lesser extent than their transformed counterparts. Thus, it seems clear that a multifaceted approach will be required to achieve specific killing of tumour cells without damage to normal cells. Utilisation of several moieties, each having tumour-cell enhanced properties in a novel MRT system should result in an overall tumour-cell specific therapeutic drug delivery system. MRTs may be engineered to include ligands (CT moieties) that are recognised by cell surface receptors that are either enriched or specific to tumour cells. For example, receptor tyrosine kinases are frequently over expressed during cancer development and are often specific to tumour types; e.g. over expression of Her-2 is associated with breast cancers.
Receptor tyrosine kinases can be targeted using single chain ligands, either in the form of anti-body fragments or as short high-affinity peptides; if these are utilised as a CT moiety within an MRT system, these should represent the first line of tumour cell specificity in a combinatorial tumour cell-killing approach. Combining tumour cell-specific receptors and CT moieties with endosomal escape (EE) and tNTS moieties would represent a tumour-selective targeting MRT with considerable potential.
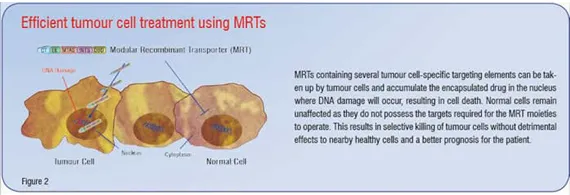
Additional modules that may act to enhance the intracellular trafficking of the MRT are also desirable. Of interest in this regard are Microtubule Association Sequences (MTASs). MTASs mediate interaction with microtubules directly or with components of the motor proteins dynein or kinesin, which mediate transport along microtubules towards the nucleus or cell periphery respectively. Several known viral and cancer-related proteins, such as p53, Rb and Rabies virus P-protein contain MTAS sequences that exploit dynein-mediated movement towards the nucleus along microtubules, resulting in increased and faster nuclear accumulation of NLS containing proteins. Incorporation of an MTAS moiety should help an MRT navigate efficiently through the crowded cytosol towards the nucleus, thus enhancing the nuclear delivery of the gene or drug of interest. In addition, it has been shown that oestrogen treatment of MCF-7 human breast cancer cells elevates dynein light chain expression, raising the possibility that upregulation of dynein light chains in tumour cells may further contribute to tumour-cell specificity of MRTs containing MTAS moieties. Figure 1 is a schematic representation of an MRT incorporating an ideal combination of targeting signals based on current knowledge. Figure 2 demonstrates the use of such MRTs to kill tumour cells effectively without damaging nearby healthy bystander cells. This results in less damage to the patient and a much better outcome after treatment.
Much remains to be done, but it would seem that achieving the ultimate goal of safe, effective and above all cell-type specific new therapies for the treatment of some of the world’s most life-threatening cancers is fast becoming a real possibility. Importantly, this seems likely to drive the pharmaceutical industry much more in the direction of efficient targeting of existing drugs to particular cell types and away from drug discovery per se.
References:
- Wagstaff, K.M. and D.A. Jans. (2007). Nucleocytoplasmic transport of DNA: Enhancing Non-viral Gene Transfer. Biochemical Journal. In Press:
- Glover, D.J., H.J. Lipps, and D.A. Jans. (2005). Towards safe, non-viral therapeutic gene expression in humans. Nature Reviews Genetics. 6(4): 299-310.
- Rosenkranz, A.A., V.G. Lunin, P.V. Gulak, O.V. Sergienko, M.A. Shumiantseva, O.L. Voronina, et al. (2003). Recombinant modular transporters for cell-specific nuclear delivery of locally acting drugs enhance photosensitizer activity. FASEB Journal. 17(9): 1121-3.
- Nishikawa, M., M. Yamauchi, K. Morimoto, E. Ishida, Y. Takakura, and M. Hashida. (2000). Hepatocyte-targeted in vivo gene expression by intravenous injection of plasmid DNA complexed with synthetic multi-functional gene delivery system. Gene Therapy. 7(7): 548-55.
- Wagstaff, K.M., D.J. Glover, D.J. Tremethick, and D.A. Jans. (2007). Histone-mediated transduction as an efficient means for gene delivery. Molecular Therapy: the Journal of the American Society of Gene Therapy. 15(4): 721-31.
- Aris, A. and A. Villaverde. (2003). Engineering nuclear localization signals in modular protein vehicles for gene therapy. Biochemical & Biophysical Research Communications. 304(4): 625-31.
- Sobolev, A.S., D.A. Jans, and A.A. Rosenkranz. (2000). Targeted intracellular delivery of photosensitizers. Progress in Biophysics & Molecular Biology. 73(1): 51-90.
- Akhlynina, T.V., D.A. Jans, A.A. Rosenkranz, N.V. Statsyuk, I.Y. Balashova, G. Toth, et al. (1997). Nuclear targeting of chlorin e6 enhances its photosensitizing activity. Journal of Biological Chemistry. 272(33): 20328-31.
- Chang, J.Y., X. Zhang, R. Komaki, R. Cheung, and B. Fang. (2006). Tumor-specific apoptotic gene targeting overcomes radiation resistance in esophageal adenocarcinoma. International Journal of Radiation Oncology, Biology, Physics. 64(5): 1482-94.
- Robert, A., N. Smadja-Lamere, M.C. Landry, C. Champagne, R. Petrie, N. Lamarche-Vane, et al. (2006). Adenovirus E4orf4 hijacks rho GTPase-dependent actin dynamics to kill cells: a role for endosome-associated actin assembly. Molecular Biology of the Cell. 17(7): 3329-44.
- Alvisi, G., I.K. Poon, and D.A. Jans. (2006). Tumor-specific nuclear targeting: promises for anti-cancer therapy? Drug Resistance Updates. 9(1-2): 40-50.
- Miron, M.J., I.E. Gallouzi, J.N. Lavoie, and P.E. Branton. (2004). Nuclear localization of the adenovirus E4orf4 protein is mediated through an arginine-rich motif and correlates with cell death.[erratum appears in Oncogene. 2005 Jun 9;24(25):4162]. Oncogene. 23(45): 7458-68.
- Danen-Van Oorschot, A.A., D.F. Fischer, J.M. Grimbergen, B. Klein, S. Zhuang, J.H. Falkenburg, et al. (1997). Apoptin induces apoptosis in human transformed and malignant cells but not in normal cells. Proceedings of the National Academy of Sciences of the United States of America. 94(11): 5843-7.
- Danen-Van Oorschot, A.A., Y.H. Zhang, S.R. Leliveld, J.L. Rohn, M.C. Seelen, M.W. Bolk, et al. (2003). Importance of nuclear localization of apoptin for tumor-specific induction of apoptosis. Journal of Biological Chemistry. 278(30): 27729-36.
- Noteborn, M.H., D. Todd, C.A. Verschueren, H.W. de Gauw, W.L. Curran, S. Veldkamp, et al. (1994). A single chicken anemia virus protein induces apoptosis. Journal of Virology. 68(1): 346-51.
- Zhuang, S.M., J.E. Landegent, C.A. Verschueren, J.H. Falkenburg, H. van Ormondt, A.J. van der Eb, et al. (1995). Apoptin, a protein encoded by chicken anemia virus, induces cell death in various human hematologic malignant cells in vitro. Leukemia. 9 Suppl 1: S118-20.
- Zhuang, S.M., A. Shvarts, A.G. Jochemsen, A.A. van Oorschot, A.J. van der Eb, and M.H. Noteborn. (1995). Differential sensitivity to Ad5 E1B-21kD and Bcl-2 proteins of apoptin-induced versus p53-induced apoptosis. Carcinogenesis. 16(12): 2939-44.
- Guelen, L., H. Paterson, J. Gaken, M. Meyers, F. Farzaneh, and M. Tavassoli. (2004). TAT-apoptin is efficiently delivered and induces apoptosis in cancer cells. Oncogene. 23(5): 1153-65.
- Poon, I.K., C. Oro, M.M. Dias, J. Zhang, and D.A. Jans. (2005). Apoptin nuclear accumulation is modulated by a CRM1-recognized nuclear export signal that is active in normal but not in tumor cells. Cancer Research. 65(16): 7059-64.
- Poon, I.K., C. Oro, M.M. Dias, J.P. Zhang, and D.A. Jans. (2005). A tumor cell-specific nuclear targeting signal within chicken anemia virus VP3/apoptin.[comment]. Journal of Virology. 79(2): 1339-41.
- Wang, Q.M., G.C. Fan, J.Z. Chen, H.P. Chen, and F.C. He. (2004). A putative NES mediates cytoplasmic localization of Apoptin in normal cells. Acta Biochimica et Biophysica Sinica. 36(12): 817-23.
- Wadia, J.S., M.V. Wagner, S.A. Ezhevsky, and S.F. Dowdy. (2004). Apoptin/VP3 contains a concentration-dependent nuclear localization signal (NLS), not a tumorigenic selective NLS.[see comment]. Journal of Virology. 78(11): 6077-8.
- Wagstaff, K.M. and D.A. Jans. (2006). Intramolecular masking of nuclear localization signals: analysis of importin binding using a novel AlphaScreen-based method. Analytical Biochemistry. 348(1): 49-56.
- Rohn, J.L., Y.H. Zhang, R.I. Aalbers, N. Otto, J. Den Hertog, N.V. Henriquez, et al. (2002). A tumor-specific kinase activity regulates the viral death protein Apoptin. Journal of Biological Chemistry. 277(52): 50820-7.
- Roth, D.M., G.W. Moseley, D. Glover, C.W. Pouton, and D.A. Jans. (2007). A microtubule-facilitated nuclear import pathway for cancer regulatory proteins. Traffic. 8: 673-696.
- Moseley, G.W., D.M. Roth, M.A. De Jesus, D.L. Leyton, R.P. Filmer, C.W. Pouton, et al. (2007). Dynein Light Chain Association Sequences Can Facilitate Nuclear Protein Import. Molecular Biology of the Cell. In press: 10.1091/mbc.E07-01-0030
- Rayala, S.K., P. den Hollander, S. Balasenthil, Z. Yang, R.R. Broaddus, and R. Kumar. (2005). Functional regulation of oestrogen receptor pathway by the dynein light chain 1.[erratum appears in EMBO Rep. 2005 Nov;6(11):1101]. EMBO Reports. 6(6): 538-44.






