
The goal of therapeutic cancer vaccines is to activate specialised cells of the immune system like cytotoxic T-cells to specifically destruct the cancer cells leaving the healthy tissues untouched. To facilitate this, the knowledge of tumour-specific antigens is required and such antigens have to be used in the right context for T-cell activation, a process referred to as vaccination. Such vaccine-induced immune responses should be effective, long-lasting and specifically directed against tumour cells. However, today's reality of cancer vaccines is still far from this point. On the contrary, the development of cancer vaccines has faced an especially difficult history so far. Like antibody-based therapies, cancer vaccines were greeted with enormous but premature enthusiasm when they showed initial impressive anti-cancer efficacy in mouse models However, when transferred into the human setting and with observation of the first clinical failures, enthusiasm turned to skepticism which remained for almost two decades.
Interestingly, therapeutic cancer vaccines have now started experiencing a renaissance. This is largely based on
- • an increasing understanding of the activation of immune cells and the requirement of immunomodulators
- • an increasing knowledge about mechanisms that allow tumours to evade or inhibit the immune cells
- • increasing possibilities to monitor vaccine-induced immune responses in vaccinated patients participating in clinical trials and utilising such information for the enhancement of the clinical design, and
- • the first successful randomised clinical trials in cancer patients that may lead to the first FDA registration of a therapeutic cancer vaccine within the next few years.
It seems that today's perception of such immunotherapeutics is rather driven by knowledge and data than mere enthusiasm and premature expectations.
Still, understanding of the immune system is not complete and for most tumour entities only few tumour-specific or tumour-associated antigens are known. Even more importantly, the relevance of the known tumour antigens is mostly unclear. Another justified point of criticism still applying to the majority of clinical trials is that the assumed relevant biological endpoints in clinical trials (vaccine-induced immune responses) firstly are not always evaluated and secondly rarely correlate with relevant clinical endpoints (objective clinical responses, prolongation of progression-free survival or overall survival).
Tumour-associated peptides
Treating cancer patients with active immunotherapy activates T-cells of the immune system specifically against the tumour cells. Tumour cells differ from healthy cells by the over-expression of tumour-associated proteins, also known as Tumour-associated Antigens (TAAs). HLA (Human Leukocyte Antigen) receptors on the cell surface display fragments (i.e. peptides) from cellular proteins to the outside, thus enabling T-cells to differentiate between healthy and tumour cells. Peptides arise from immature translation products and ageing proteins. Inside living cells, peptides are bound to HLA receptors and shuttled to the cell surface. These peptides that are presented by the tumour cells are called Tumour-associated Peptides (TUMAPs).
There are two classes of TUMAPs. HLA Class I TUMAPs are short peptides (8 to 10 amino acids) that activate Cytotoxic T Cells (CTLs). Activated CTLs can directly kill tumour cells by secreting cytolytic substances, or by driving tumour cells into apoptosis. HLA Class II TUMAPs are slightly longer peptides (approximately 15 to 25 amino acids) that activate T helper cells. Activated T helper cells provide help to CTLs by locally increasing the concentration of certain cytokines. T helper cells can also have direct anti-angiogenic effects on the tumour, suppressing the growth of new blood vessels. The platform described in this article allows discovery and development of both Class I and Class II TUMAPs.
Figure 2: New Novel Technologies for Vaccine Development
• Safe: Various TUMAPs (mainly in the field of melanoma vaccination) have been administered in thousands of patients and were tolerated very well.
• Scalable: Peptide synthesis is a robust and semi-automatic chemistry in completely cell-free system without the requirement of any recombinant expression. Manufacturing up to the kilogram range is feasible at low costs compared to manufacturing of proteins.
• Stable: Peptides as well as peptide mixtures can be lyophilised and have been shown to be stable when stored aT-20?C or 4?C for years. They can be shipped at room temperature conditions.
• Pure: Similar to small molecules synthetic peptides can be manufactured at very high purities excluding any tolerising cellular antigens and normal proteins found in all cellular and autologous approaches, thus avoiding the inclusion of autoantigens.
• Processed: In contrast to protein-based vaccines TUMAPs can be bound directly by antigen processing cells, such as dendritic cells in the skin, which subsequently can activate na?ve T-cells. No further antigen processing by the Antigen Presenting Cell (APC) is required.
• Specific: The activation of T-cells by TUMAPs is a highly specific process. Additionally, TUMAPs, in sharp contrast to monoclonal antibodes (mAbs), give access to tumour antigens irrespective of the intra or extra-cellular location of the target allowing a more careful selection among a much larger pool of antigens. mAbs can only recognise target antigens expressed either as soluble factors (e.g. Vascular Endothelial Growth Factor VEGF) or as surface-standing molecules (e.g. VEGF receptor).
• Evaluable: TUMAPs are not only used for vaccination but are also of use in the direct read-out of immunological responses to specific antigens, an important parameter in early-stage clinical development.
• Combinable: Pharmaceutical development and GMP manufacturing of mixtures of 10 or more peptides is feasible allowing to create defined multi-target vaccines.
• Matching regulatory requirements: TUMAPs match expectations of regulatory authorities, i.e. clean technology, exactly defined compounds, clear characterisation of all compounds and impurities.
XPRESIDENT - Identification, selection and validation of relevant TUMAPs from cancer tissues
XPRESIDENT, a technology platform originally created in the laboratories of the pioneering immunologist Hans-Georg Rammensee and colleagues at the University of Tuebingen (Germany), and continuously enhanced at the spin-off immatics biotechnologies.
XPRESIDENT is the first platform that allows:
1. the identification of large numbers of naturally presented novel HLA-binding peptides directly from primary tumour tissues
2. selection of tumour-associated peptides by differential gene expression and peptide presentation analysis and
3. validation of selected candidates by measurement of in vitro T-cell responses.
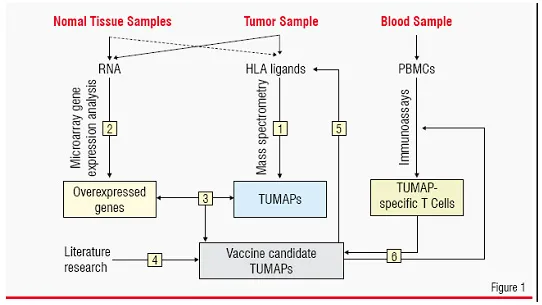
This approach combines methods from genomics, proteomics / peptidomics, bioinformatics and T-cell immunology. Using this technology, off-the-shelf cancer vaccines consisting of multiple tumour-associated peptides can be developed. Samples from surgically removed malignant and normal tissue from patients and blood from patients or healthy donors are analysed in a stepwise approach (Figure 1):
1. HLA-bound peptides from the malignant material are isolated and identified (i.e. sequenced) with high sensitivity using latest technologies for on-line chromatographic separation and high-end mass spectrometry on the nano scale.
2. Genome-wide mRNA expression analysis by microarrays is used to identify genes over-expressed in the malignant tissue compared with a range of normal organs and tissues.
3. Identified peptides are compared to gene expression data. Peptides encoded by selectively expressed or over-expressed genes as detected in step 2 are considered suitable candidate TUMAPs for a multi-peptide vaccine.
4. Literature-knowledge is used for additional evidence supporting the relevance of the identified peptides as TUMAPs.
5. The relevance of over expression at the mRNA level is confirmed by redetection of selected TUMAPs from step 3 on tumour tissue (as well as normal tissue for one selected TUMAP) and optionally by quantification of the TUMAPs themselves.
6. Peripheral T-cells of patients or healthy individuals are tested for reactivity against the tumour-associated HLA ligands using several standardised immunoassays (in vitro T-cell assays).
The first major advantage of this approach is that resulting TUMAPs are confirmed to be naturally processed and naturally presented in real tumours reflecting the actual in vivo situation in cancer patients. This has several advantages over other technologies relying on cell lines or computer algorithms used for prediction of putative TUMAPs. Cell lines are known to be very distinct from primary tumour material because they display different gene expression patterns and antigen profiles compared to primary tumours. For instance, this is the case when tumour associated antigens have relevant functions for interaction with the extra-cellular matrix. Also, cell lines may use different antigen processing rules (e.g. different proteasomal species). Furthermore, cell lines reflect only one type of tumour cell out of a larger population of cancer cells. When looking at primary cancer tissue, it is also possible to look at different cell types within tumour samples, such as tumour-supporting stroma cells and endothelial cells of tumour vascularisation.
The second major advantage of this platform is the identification of a relatively large number of novel tumour antigens. This allows developing peptide-based vaccines based on multiple TUMAPs (typically 10 or more TUMAPs and each TUMAP known to be presented by at least 50% of analysed tumour tissues) in one single product even for tumour entities where only few antigens have been known so far. The advantages of using multiple peptides are:
No tumour-associated antigen is present in every tumour. With every additional antigen used there is an increased chance that the vaccine will reach its target or even several relevant targets simultaneously.
Priming of only one kind of cytotoxic T-cell(CTL) with one TUMAP is usually insufficient to eliminate all tumour cells. Tumours are very mutagenic and are thus able to respond rapidly to CTL attacks by changing their pattern of expressed proteins, allowing them to escape from the recognition by CTLs. Several activated T-cells can act synergistically by simultaneously attacking a corresponding number of independently encoded tumour antigens reducing the chances of a tumour cell to evade the immune response by down-regulating single targets.
IMA901 - The first product generated with XPRESIDENT
The HLA peptidome of 32 primary renal cell carcinoma samples routinely resected from patients was systematically investigated using the XPRESIDENT technology. 9 HLA-A*02- and 1 HLA-DR- restricted TUMAPs, derived from nine different tumour antigens were selected. All selected TUMAPs were confirmed to be immunogenic in vitro, although immunogenicity varied between single TUMAPs. This mixture was pharmaceutically developed to a stable lyophilised formulation that is rapidly dissolved in physiological buffer and ready for intradermal administration in a single shot. The product was designated IMA901. In the first-in-man trial, one additional well described viral peptide derived from Hepatitis B virus was included into IMA901 as a marker.
First proof-of-principle in the clinical setting
Twenty-eight HLA-A*02-positive stage III/IV RCC patients were enrolled in a single arm, multi-centre study and received eight intradermal vaccinations each consisting of IMA901 (including the HBV-derived viral marker peptide) and GM-CSF as immunomodulator. T-cell responses were measured systematically in peripheral blood using IFN-gamma ELISPOT and HLA multimer analysis and CD4+ Foxp3+ regulatory T-cell (Tregs) levels.
IMA901 rapidly induced T-cell responses in 75% of evaluable patients (N=27). T-cell responses were detectable already within the first 14 days after the first vaccination, peaked subsequently and were sustainable until follow-up in the majority of patients. The overall tumour assessment in patients with measurable disease (N=23) revealed that eight patients (35%) demonstrated a clinical benefit (one partial response and seven stable diseases). Most importantly, patients eliciting multiple TUMAP responses-observed in 30% of patients -showed a higher clinical benefit rate. This correlation was found to be statistically significant (p=0.018).Furthermore, another correlation was observed: patients with lower levels of so-called regulatory T-cells at study onset showed a higher proportion of immune responses to multiple TUMAPs (p=0.016). Such CD4+ Foxp3+ regulatory T-cells (Tregs) have been recently described as key mediators of peripheral immune tolerance and may be important inhibitory cells against immunotherapeutics in vivo. Importantly, both correlations were only observed for T-cell responses to TUMAPs. However, no correlation of HBV marker peptide responses to either Tregs or clinical benefit was observed.
Outlook
Although conclusions from this pilot trial should be treated with caution due to the small study size and short duration of clinical observation, results seem remarkable due to the fact that most immunotherapy trials have failed to demonstrate correlations between immune responses and clinical benefit. It can be speculated that this may be due to two important characteristics of IMA901: the use of multiple peptide antigens as well as the use of naturally presented peptides. Multi-centre randomised clinical trials are currently conducted in Europe with the goal to demonstrate the clinical benefit of IMA901 in a larger study population treated and observed for a longer duration.
Additionally, the XPRESIDENT platform is not limited to renal cell carcinoma but has also been successfully applied to other tumour entities. Further, multi-peptide product candidates will soon be introduced into clinical development. It remains to be seen whether this novel class of naturally presented antigens will be able to make a difference to cancer immunotherapy. The first results have provided some hope-a hope driven by data and new knowledge.
Read the full article — it's free
Register with Pharma Focus Asia to unlock expert insights, research articles, and in-depth industry analysis.






