In this article we will discuss regulatory recommendations concerning the determination of process-related impurities during biopharmaceutical drug manufacturing. Furthermore, we suggest their implementation into the setup of a reliable Host Cell Protein (HCP) monitoring using Enzyme-linked Immunosorbent Assay (ELISA) during early and late stages and provide valuable insights into critical steps along the way of HCP ELISA setup.
Biopharmaceutical drugs make up a large portion of global pharma sales, with 8 of the top 10 global drug blockbusters in 2019 being recombinant biopharmaceuticals. Drug development is separable into five phases: i) Pre-clinical Phase, ii) Clinical Trials Phase I, iii) Clinical Trials Phase II, iv) Clinical Trials Phase III, and v) the Market Authorisation Application (MAA) followed by the Drug Launch, after all previous steps have been passed successfully. To ensure high-level patient safety during Clinical Trials and upon drug product release, a multitude of regulations have been authored by different regulatory bodies, such as the US FDA and the EMA. The MAA requires a profound overall assessment of potential risks and benefits, the so-called Critical Quality Attributes (CQA), which are included in the Common Technical Document (CTD). Host Cell Proteins (HCP) are one such CQA, they stem from the production cell line used for biological product manufacturing. Defined as process-related drug impurities, HCP can negatively influence the quality, safety, and efficacy of a biological drug product. The HCP formation by such complex cellular production systems is influenced by a multitude of biotic and abiotic factors, which makes it hard to predict the HCP pattern of individual manufacturing processes. In particular, ICH Guidelines Q6B, Q8(R2) and Q11 define such impurities and address the need for the precise monitoring and the reduction of HCP during stepwise Downstream Processing (DSP), all the way down to low amounts. Although no precise values are specified, the common agreement is to reduce the HCP burden below 100 ppm in the final drug substance. Naturally, the accurate detection of HCP impurities in subsequent DSP samples down to the final drug substance heavily depends on the establishment of a reliable and robust method for HCP measurement. To achieve this, the use of multi-faceted HCP analysis methods is recommended. The Enzyme-Linked Immunosorbent Assay (ELISA) is still considered the gold standard for HCP measurement, as it has advantages such as high speed, sensitivity and high throughput. Nevertheless, to overcome intrinsic limitations of individual methods for HCP quantitation, the implementation of orthogonal methods is strongly advised. This will be addressed below.
Selection of the Appropriate HCP ELISA
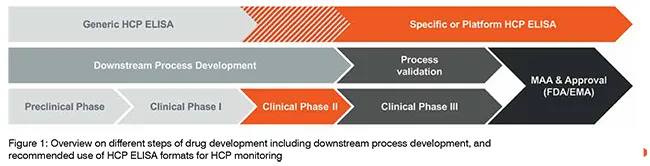
The HCP ELISA for HCP quantitation can be divided into three main formats: i) the commercial ELISA, also referred to as generic HCP ELISA; ii) the platform (or multi-product) HCP ELISA; and iii) the process-specific HCP ELISA. While the generic HCP ELISA makes use of a broadly active antibody coverage approach which is specific only for the selected cell line of recombinant protein production, the latter two HCP ELISA formats are based on greater specificity towards the manufacturing and processing of particular biopharmaceuticals. The answer to the question: “Which HCP ELISA is best to use during which phase of development of a drug candidate?” is not a clear dichotomous one, however there are common agreements for when the usage of the comprehensive HCP determination of either one or the other format is favoured. When discussing the advantages and disadvantages of each format, one needs to recall the previously-mentioned steps of drug development and all accompanied processes. During Phase I of clinical trials, the drug substance is required to be produced under GMP-grade manufacturing conditions at pilot scale volume and relies on proper technology transfer from pre-clinical process development, with putative changes in DSP still likely to happen. The drug substance used during Phase II of clinical trials has to be produced at larger volumes than the pilot-scale volume, using defined process parameter specifications, and thus meeting the requirements for high process robustness. By contrast, the drug substance used for testing in Phase III of clinical trials has to meet identical requirements as for continuous drug production after marketing, providing at least three different batches of drug substance at production scale. For this phase of drug manufacturing, all of the processes need to be validated, including the use of analytical methods. If changes to manufacturing steps are carried out at this stage, process validation has to be repeated until a sufficient consistency is achieved. This also includes the monitoring of residual HCP impurities, which can technically vary in amount and composition when changes during manufacturing or DSP are introduced. The common recommendation is therefore to rely on a broadly active generic HCP ELISA only during method development. When moving forward towards application for extended clinical trials in Phases II and III, the implementation of a process-specific HCP ELISA usually proves adequate for HCP monitoring, allowing the criteria for assay validation to be meet. The use of a platform HCP ELISA can be sufficient when manufacturing and DSP conditions and the principle nature of different biopharmaceuticals only vary in a small range without having major influence on the respective HCP pattern (see also Figure. 1). One aspect to factor in when being confronted with the decision of which HCP ELISA format to use for continuous HCP monitoring is the potential risk of limited antibody availability. While a commercial HCP ELISA might not be supplied constantly during the average life cycle of a biopharmaceutical, this risk can be significantly mitigated when choosing the development of a process-specific HCP ELISA with a large-scale immunisation approach (see below).
Careful Functional Assay Design
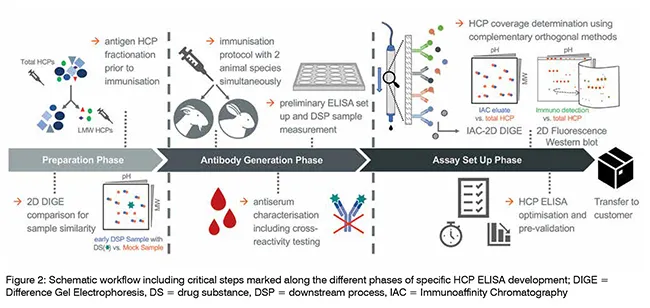
The suitability of an HCP ELISA for HCP monitoring is usually determined by performance criteria, such as the assay’s sensitivity to detecting HCP trace amounts even in highly purified samples demonstrating the HCP logreduction over the various steps of DSP, the stringent dilution linearity, and a sufficient HCP-specific antibody coverage. In the following, critical aspects during specific HCP ELISA design are highlighted (see also Figure. 2). One such factor is the selection of appropriate HCP mock material for polyconal antibody (pAb) generation. Ideally, a great similarity in the HCP spectrum of both the HCP mock material and a process sample originating from recombinant drug substance (DS) production would indicate mock suitability for pAb generation. This can be analysed with Sodium Dodecyl Sulphate Polyacrylamide Gel Electrophoresis (SDS PAGE) using two-dimensional Difference Gel Electrophoresis technology (2D DIGE). Here, the HCP spot pattern of an early DSP sample is qualitatively compared to the pattern of a given mock sample. Also, HCP of low molecular weight (LMW) tend to be less immunogenic, and thus an underrepresentation of LMW-specific antibodies is frequently observed in standard HCP immunisation regimes. A counterstrategy is the fractionation of the HCP prior to the immunisation of host animals, with both fractions in parallel. Furthermore, a differential pAb panel can be generated by employing two different host animal species (goat and rabbit) to be immunised, which allows for another level of selection. The HCP-specific immune response in individual animals is monitored by ELISA titer determination and Western blotting. This also includes antiserum testing for cross-reactivity against the DS, in order to exclude false-positive HCP ELISA results during the process sample analysis. A preliminary ELISA then is set up with affinity-purified antibodies and its performance is evaluated based on the key parameters detailed above. From this, the species which matches best the quality criteria is selected for extended immunisation and subsequent large-scale antibody purification.
Take Home Messages
- Ensure HCP monitoring along the biopharmaceutical drug production process
- Allow sufficient development time for a customised HCP ELISA assay
- Use separate LMW HCP fraction for immunisation to tackle immunogenicity issues
- Perform immunisation of two different species for optimal results
- Consider methodological limitations and increase the reliability of your HCP coverage determination by applying orthogonal methods such as 2D Western blotting, IAC-2D DIGE and potential supplementary mass spectrometry
Reagent Characterisation by Use of Orthogonal Methods for HCP ELISA Qualification
The assessment of the suitability of an HCP ELISA for HCP monitoring includes the HCP-specific antibody coverage. This coverage analysis is performed to determine the ratio of HCP species that are successfully detected by the pAb, expressed as percentage HCP coverage. The traditional method is the 2D Western blot, using the HCP mock material and/or an early DSP sample. As mentioned above, the use of orthogonal approaches is advised. One such method involves the usage of Immunoaffinity Chromatography (IAC) with immobilised pAb, followed by 2D DIGE. The 2D DIGE comparison of the IAC eluate of the pAb-bound HCP fraction with the total HCP sample allows for the estimation of HCP coverage by the antibodies under non-denaturing conditions. However, both analytical approaches come with inevitable methodological limitations. To achieve a scientifically sound estimation of the HCP coverage, the application of both methods for best reagent characterisation is strongly recommended. The completion of HCP ELISA set up includes optimisation with a focus on the titration of all reagent concentrations and incubation times, accompanied by an evaluation of assay specificity, accuracy and precision. This state-of-the-art HCP assay development includes the suggested use of both the HCP mock material and a relevant process sample during method optimisation. When factoring in the average developmental time for a process-specific HCP ELISA of at least 1.5 years, manufacturers of biopharmaceuticals are advised to begin planning for the introduction of reliable HCP monitoring assays during drug development as early as possible, in order to have a functional HCP ELISA at hand when assay validation is due. This also plays a role when a switch of manufacturer for continuous drug supply is considered. Taken together, the consideration of the critical steps mentioned here allows for the implementation of robust and reproducible HCP monitoring during biological drug manufacturing.
Suggested Reading:
Bracewell DG, Francis R, Smales CM. The future of host cell protein (HCP) identification during process development and manufacturing linked to a risk-based management for their control. Biotechnol Bioeng. 2015;112(9):1727-1737. doi:10.1002/bit.25628
Vanderlaan M, Zhu-Shimoni J, Lin S, Gunawan F, Waerner T, Van Cott KE. Experience with host cell protein impurities in biopharmaceuticals. Biotechnol Prog. 2018;34(4):828-837. doi:10.1002/btpr.2640
Zhu-Shimoni J, Yu C, Nishihara J, et al. Host cell protein testing by ELISAs and the use of orthogonal methods. Biotechnol Bioeng.2014;111(12):2367-2379.doi:10.1002/bit.25327
F. Wang, D. Richardson, and M. Shameem, “Host-Cell Protein Measurement and Control” BioPharm International 28 (6) 2015
USP 39 Published General Chapter <1132> Residual Host Cell Protein Measurement in Biopharmaceuticals
ICH Guideline “Specifications: Test Proceedures and Acceptance Criteria for Biotechnological/Biological Products Q6B” (1999)
ICH Guideline “Pharmaceutical Development” Q8(R2) (2009)
ICH Guideline ”Development and Manufacture of Drug Substance (Chemical Entities and Biotechnological/Biological Entities)” Q11 (2012)
ICH Topic M 4 Q Common Technical Document for the Registration of Pharmaceuticals for Human Use – Quality (CPMP/ICH/2887/99) (2003)










