
REGULATORY REFORM
How China is investing in its pharmaceutical market
Mingping Zhang, Vice President of Technology PAREXEL Regulatory Consultant
The nuances of clinical and regulatory product development in China, and its similarities and contrasting aspects in comparison to the rest of the world are discussed in this article. The specifics within risk management and the global regulatory authorities that contribute to this complex landscape were also explained.
Recent legislation has forced the Chinese regulatory environment to evolve, establishing a friendlier environment for pharmaceutical development. These changes may opendoors for pharmaceutical companies, but they also create the need to adapt to the fast-changing regulatory requirements and the uncertainty that comes with legislative change.
The root cause of these changes lies in China’s economic growth. China is set to have the largest middle-class group in the world, which will significantly alter the needs of people and as a result, reshape the market. China’s healthcare spending in 2016 was four times the level of 2008 – making it no surprise that experts anticipate China will become the second largest pharmaceutical market by 2020.
Catalyst for Change
China’s economy needed a reboot and the healthcare and pharmaceutical industries were key to leading the growth. However, compared to many other areas, the pharmaceutical industry is one of the weakest in China due to extremely long and unpredictable regulatory timelines. To help address some of these issues within the industry, the Chinese government issued a series of regulations and reorganisations that aim to make fundamental changes to the Chinese regulatory administration system. The aim was to cater to the needs of the Chinese people and encourage pharmaceutical development.
Chinese Regulation Timelines
2015: The State Council initiated the China regulatory reform.
2017:In October of 2017, China CPC Central Committee & State Council made another important announcement of regulatory reform.
2018: Based on the decision by the 13th National People's Congress, the Chinese government was reshuffled in March of this year. Since this change, the China State Council consists of 26 ministries and commissions in addition to the General Office of the State Council.
With this shift in power there was a reorganisation of the pharmaceutical industry. To start, the State Market Regulatory Administration (SMRA) was established and set to report directly to the State Council. Furthermore, the China National Drug Administration (CNDA), formerly the China Food and Drug Administration (CFDA), will report to the SMRA.
The Resulting Reforms
The efforts of the Chinese government over the past few years has resulted in substantial change throughout the industry. From protocols, to access, to speed-to-market, the reform of the Chinese pharmaceutical industry has affected the policies and structures of organisations across the pharmaceutical continuum.
Policies
In April 2018, Premier Li Keqiang proposed several policies for imported drug products to China. These policies include the following:
• Since May 1 of this year, there are zero tariffs on oncology products imported to China, including new products and generics.
• Government and industry leaders also announced on May 1a new regulation that reduced the value-add tax (VAT) on oncology pharmaceutical products (API & DP) from 17 per cent to 3 per cent.
• Based on CFDA Order No 12 (2018), all the import chemical products are exempt from sample testing for every import batch, except the first import batch (risk-based testing may require for later batches) effective April 24, 2018.
• The Chinese government added 44 high-priced drugs to the National Reimbursement Drug List (NRDL) – a sign that China is expanding its coverage and improving Chinese patient benefits.
Regulatory Reviews
Other efforts by the government include expedited reviews for urgently needed therapies. If drugs and devices offer new solutions for treating life-threatening diseases or address critical unmet medical needs, they can be eligible for conditional approvals. This holds true as long as early and mid-stage study data indicate their efficacy and predict their clinical value.
This initiative not only shortens regulatory approval time, but also lowers the regulatory approval requirements. For some drugs, companies will have chance to use foreign data to support Chinese market access approval directly with post approval commitment. Even if the data package falls in to the ‘partially accept’ category after the ethnical sensitivity analysis, foreign data could be admissible.
Drug Access
One of the most aggressive approaches was announced in April 2018, allowing certain hospitals in Hainan province to import drug or medical devices for unmet medical needs without CNDA review and approval. The clinical data collected in these hospitals could support Chinese registration if it can meet certain criteria. This kind of policy not only gives Chinese patients the chance for early access and advanced therapy, but also gives more flexibility for Chinese development.
In order to protect patients, however, the Chinese Court has endorsed a death penalty for falsification of drugs. Before regulatory reform, the most serious punishment for this kind of crime was that companies were not allowed to submit any new submission for three years.
The government also expanded drug access to investigational products. Patients not enrolled in a clinical study may have access to investigational products if they give informed consent and if the investigational product is intended for treating life-threatening diseases with no effective treatments. The safety data generated from this expanded use can be used to support marketing authorisation applications.
Stricter Approval Criteria
One of the measures taken by the CFDA includes establishing stricter approval criteria. This implies that CNDA will implement internationally aligned requirements. With this, CNDA also joined the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) in 2017 and 5 of ICH’s guidelines were officially implemented in China based on CFDA Order NO 10 in 2018. Up to now, CNDA published about 120 guidance and translated over 500 WHO, ICH, FDA, EMA guidance. It is mutually agreed that global guidance can be referenced if certain specific Chinese guidance is not available.
Documentation
In addition, the ‘China DMF’ is implementing a rule which requires companies handling API, excipients and package materials to file their Drug Master File (DMF) document to CNDA. This API DMF document should follow the CFDA Order 80, 2016 requirements – noting the application dossier requirement for the new classification of the chemical drug. Similarly, the excipients and package materials DMF should follow the CFDA Order 155, 2016 requirements – noting the application dossier requirement for the pharmaceutical package material and excipients.
Addressing Drug Lag
In March 2016, the Chemical Drug Registration Category changed based on CFDA order NO 51 (2016), which aims to solve the drug lag and encourage innovation in China. For generic medicine, the approval criteria has changed from ‘meet specification’ to ‘demonstrate the similarity in both quality and clinical performance.’ For innovative drugs, the definition has evolved from ‘never marketed in China’ to ‘never marketed anywhere.’ New drugs will receive the ‘data protection,’ which is the extra regulatory benefit that CNDA offers.
Implementing Data Protection
On April 26 2018, CNDA announced public consultation for the new draft regulation of Drug Trial Data Protection. During the data protection period, CNDA will not approve any other sponsors which intend to refer to the originator’s label, unless it generates all original clinical data or gets the originator’s authorisation. Innovative chemical drugs, orphan drugs or paediatric drugs with non-medical needs in China may be awarded up to six years of data protection –innovative therapeutically biologics may be awarded up to 12 years of data protection. One of the key considerations of the data protection period is whether China has been integrated into the global synchronised development – one of the key criteria to receiving the longest protection period. Additionally, the data protection period will depend on the length of the drug lag in China – the protection period will be deducted from the China drug lag time.
Expanding Trial Sites
Data protection is just part of the ongoing effort by the CNDA to create a friendly pharmaceutical development environment in China. There are about 1.4 billion people in China; as a result, the ability to quickly recruit patients is one of the major advantages of running clinical trial in China. Prior to the reform, only 619 certificated clinical sites were allowed to perform clinical trials. With the China regulatory reform, all GCP compliant hospitals are allowed to perform clinical trials without certificate. The ‘hospital’ here refers to over 2000 Grade III hospitals (tertiary referral centres), over 8000 Grade II hospitals and over 20000 Grade I hospitals. In addition, the hospitals and principle investigators are incentivised to perform clinical trials because clinical research receives credit and counts towards KPI.
Speed of Process
It used to take two to three years to get the Clinical Trial Application (CTA) in China and the average start-up timeline is four to six months. To start a clinical trial in China, sponsors need to go through the sequential process: CTA (two to three years), Ethnical Committee (EC) approval (one to three months) and Human Genetic Resources Administration Committee (HGRAC) approval (two to three months), before the first patient even enters through the doors. After reform, the CTA & EC can go in parallel, and the HGRAC can also go through the partially parallel process – expediting the process significantly. This action is expected to reduce HGRAC approval time to one to two weeks.
In addition, the CTA approval timeline has been shortened to six months in 2017 and it will drop to 60 working days in 2018. Companies used to get EC approval from each clinical site, which was bottle necked by non-standardised protocols. Now the new regulation encourages a central/regional ethical committee, which will help to simplify and standardise the EC approval process. Ideally, CTA & EC approval goes in parallel which cost 3 month, then HGRAC approval and site initiation for 1 month; the total timeline from submission to First Patient In (FPI) could be four months in 2019.
Not only has the timeline shortened for Chinese trials, but certain technical requirements have helped smooth the Chinese trial startup process. CNDA used to require comprehensive CMC information even for early phase clinical trials. Now the draft Phase I and Phase III application guidance standardises requirements on different phase clinical trial applications. Similarly, mandatory sample testing used to be the holdup of CTA approval, especially with bio-products. After reform, the sample testing will be risk-based for most products.
Additionally, regulators established the measure to ‘accelerate approval for innovative drugs.’ The NDA approval timeline used to be two to three years, but after reform, the new NDA timeline could be 120 working days for accelerated approval and 200 working days for standard approval. Additionally, foreign clinical data could support Chinese registration if the efficacy and safety are adequate and could extrapolate to Chinese patients. Ethical sensitivity should be well addressed, and all the clinical sites must comply with ICH GCP requirements and should be ready for CNDA inspection.
Improved Data Integrity
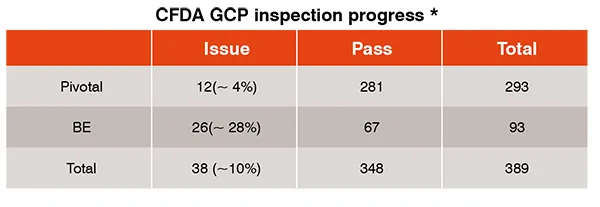
Clinical trial data integrity use to be a concern of pharmaceutical companies and health authorities. As a result, CFDA initiated the China GCP inspection for all the Market Authorized Application (MAA) in 2015 by implementing the ICH aligned GCP requirements. About 70 per cent of clinical trials which were performed under less strict GCP environments have been withdrawn. The rest of the applications need to go through GCP inspection. Table 1 provides the inspection results, which imply that China clinical sites are capable of generating high quality clinical data and fully complying with ICH GCP requirements.
As the Chinese pharmaceutical market expands to be one of the top markets in the world, regulators need to keep pace and facilitate growth. With reforms that address the biggest pain points of the industry, China will be able to embrace technology and international data to improve processes and ultimately enable more scientific discoveries to help patients.
Author Bio

Zhang has more than 15 years of regulatory experience in the pharmaceutical industry and health authority. He currently serves as the Vice President (Technical) in PAREXEL Consulting and is mainly responsible for regulatory strategy and CMC consultant in China. Previously he was the CMC Director at BeiGene where he was responsible for development relevant CMC activities; including drug substance & drug product manufacturing, clinical supply & regulatory CMC. He has previous experience as a DRA CMC Manager and supported the CTA, NDA, supplemental application, variation management, and other regulatory relevant CMC issues for Novartis China. Zhang received his Master’s Degree in Chemical Engineering from the National University of Singapore and his Bachelor’s Degree in Biochemical Engineering from Beijing University of Chemical Technology.




