
The year 2004 marked a 20-year low in the introduction of new medical treatments into the global market. Over the last two-and-a-half decades the pharmaceutical industry has been under increasing pressure to constrain its costs and expenditure whilst the pressures for replenishing the dwindling drug development pipeline have remained unabated.
Though cost is usually cited as the main reason to explain this trend, one must also consider other factors like increasing regulations, generic competition, reimbursement challenges, development models, recruitment and retention.
In the past, the pharmaceutical industry has witnessed consolidation through mergers and acquisitions which has led to the formation of mega giants, e.g. Pfizer and AstraZeneca. This period of consolidation may have initially led to some cost efficiencies and economies of scale, nevertheless it has failed to address the underlying reasons for the lack of organic growth of the drug development pipeline.
More recently, Big Pharma has concentrated its efforts on biotechnology companies which offer the promise of new discovery and development platforms. The increasing trend of mergers and acquisitions of biotechnology companies has spawned a new term for the industry “Biopharmaceuticals”.
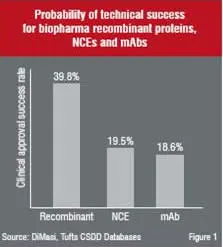
The success of Biotechs can be illustrated if one was to compare the clinical approval success rates of biologics (recombinant proteins and monoclonal antibodies) against new chemical entities (Figure 1).
Although the success factors for biologics appear to be more favourable as a whole, the challenges listed earlier still pose a formidable obstacle for developing biotechnology and pharmaceutical companies. Prior to embarking on a development programme one should therefore investigate these challenges further in order to fully appreciate the complexities of the development process.
Drug development process
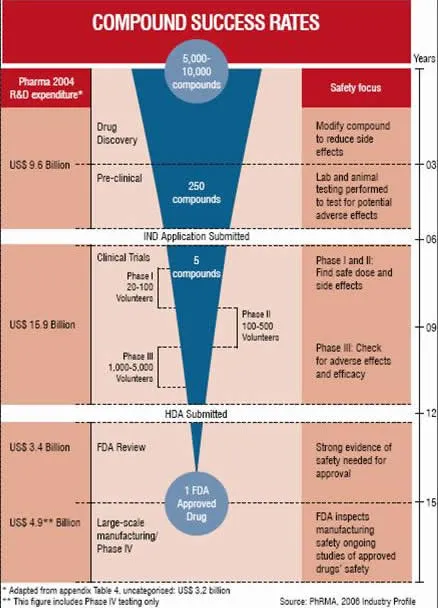
The overall development approval process is complex and involves lot of time as per the requirements of US FDA guidelines. The process for a new compound from synthesis to obtaining marketing approval can take from anywhere between 10 to 20 years, with an estimated average of 9 to 12 years.

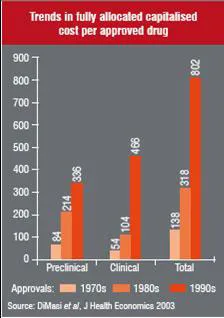
Over the last two-and-a-half decades the length and cost of this process has increased considerably. It is important to understand at what stage of development expenses occur. Figure 2 illustrates the cost of this process for the period of three decades 1970s-1990s and the following points can be deduced from it:
• Total drug development costs have risen almost six times
• Clinical costs have risen at a faster rate at almost nine times over the last three decades
The development process of a new compound is initiated at the pre-clinical stage where a compound undergoes trials in vitro and then in vivo in laboratory animals to evaluate the pharmacological and toxic effects. The drugs genotoxicity is also investigated at this stage.
At this point if the compound under investigation is worth pursuing, an Investigational New Drug (IND) application will be filed with the US FDA. The IND will document the results of short-term toxicity testing in at least two animal species and also describes the compound’s pharmacological profile.

The FDA has 30 days to review the submitted IND application. If the FDA does not place a hold on the application, the drug developer can begin “first in man” Phase I clinical trials in humans.
Clinical (human) testing usually proceeds through three successive clinical phases prior to a marketing application followed by post-marketing Phase IV trials.
The costs associated with each clinical phase increases progressively from Phase I through to Phase III.
The largest single investment throughout development will be at Phase III due to the clinical trial sample size which usually ranges in the thousands but also due to the design of the study as it must include a comparator arm(s). Sourcing of the comparator drug(s) is usually an expense which is paid for by the developer. However, this is not always necessary as in some countries the comparator(s) may be the standard of care and as such may be provided by the healthcare provider.
Challenges
Regulatory requirements and commitments have increased progressively over time and this has led to an increase in both the trial size and the length and hence a rise in the overall cost of the development process. More subjects are needed as many of the treatments under development require greater numbers to achieve FDA statistical standards for demonstrating safety and efficacy. Clinical trial sample sizes increased at an average rate of 7.47 per cent per annum, from the 1970s to 2001, as reported by DiMasi et al.
As a result of the increasing clinical trial sample sizes, it has become necessary to conduct trials in many countries, each with their own unique regulations.
One must also consider the number of existing guidelines which have been introduced progressively over the years, e.g. approximately
• Thirty ICH guidelines relevant to global drug development
• Seventy FDA guidelines relevant to clinical research
• Six guidelines that support the EU clinical trial directive.
More recently there has been an increase in the number of safety recalls of previously approved drugs, e.g. cox inhibitor / Vioxx. This has led to an emphasis by regulators on the
development of risk profiles for pharmaceutical products and also closer scrutiny of clinical trials combined with post-marketing commitments.
Other requirements include the electronic submission to the regulatory authorities which is mandatory in some key countries like the US. In the EU, the Electronic Common Technical Document (eCTD) will soon be the mandatory format for EMEA submissions and is already compulsory for some national authorities. The transition to eCTD has many benefits including better lifecycle management of regulatory submissions and easier review of documents which should reduce the review time. Developing companies must quickly adopt this approach as they need to be eCTD-ready in order to actively manage the product lifecycle during the development process.
Reimbursement
Obtaining approval to market a compound is sometimes only half the battle won especially in Europe where additional health economic data is usually required in order to ensure reimbursement from health authorities. Tools (i.e. Patient Reported Outcomes or PROs) should be utilised and planned into clinical trials early on in the development process so as to ensure maximum reimbursement. Innovative target oncology treatments (i.e. Bevacizumab, sorafenib, sunitinib and temsirolimus) have not been granted approval for reimbursement by the UK National Institute for Health and Clinical Excellence (NICE). Following a preliminary review, NICE ruled that although these drugs are clinically effective, they do not provide good value for money (the cost of these treatments are approximately US$ 5,000-10,000 per month).

Generic competition
It is no longer acceptable for drug developers to commence efforts to extend a product’s lifecycle as patent expiry approaches. When a drug’s market exclusivity is lost, a meltdown in sales may be experienced within one business quarter. Companies need to be developing a strategy to protect against patent expiry even before a product is launched. There are numerous potential strategies and the choice depends on the particular circumstances of the product. For example, second-generation versions with improved safety or efficacy, new formulations, combination products, licensed generics, and Over-The-Counter (OTC) switching, are all the potential choices.
Recruitment and retention
Competition from similar studies run by rival companies combined with increasing clinical trial sample sizes has led to expanding recruitment efforts to more exotic countries. This in itself brings its own complications due to the uncertainty associated with unfamiliar regulations but sometimes it is worth the extra risk as recruitment rates tend to be higher than the more established countries.
As clinical trial designs become lengthier and complex, the burden on subjects increases and hence the retention rate decreases. Overcoming this problem is the key in keeping the costs down as subjects who withdraw from the study will usually need to be replaced. Recruitment can be further compounded by regulatory requirements to include certain patient populations (i.e. children).
Development models
In-house development is usually the preserve of large and medium-sized pharmaceutical companies that have the money to build large research teams. The advantage of this model is that you are not always reinventing the wheel and it results in continuity, leading to expertise and cost-efficiency. The industry has moved away from this trend mainly to streamline existing teams and save costs. However, this is not an option for developing biotechnology and pharmaceutical companies as it is hugely expensive.
Outsourcing to a large Clinical Research Organisation (CRO) has many advantages and is usually the option that developing companies opt for, but at the same time this model does have some disadvantages which must be thoroughly considered before this option is selected. Any wrong decision for a company with limited resources can potentially be catastrophic.
Solutions
Alternative development model
An alternative development model that is more cost and time efficient may be to partner with a consultancy service which can assist the project by managing the whole development process or just a small portion of it.
The advantages of this type of model are:
• The consultancy only utilises specialist niche service providers which helps focus on time critical components without sacrificing quality whilst keeping cost to a minimum
• Smaller teams, sometimes limited to 2-3 people, ensure continuity through lower turnover
• Ongoing training and mentoring may be provided enabling the company to establish or strengthen its own R&D team where experience may be limited
• Implementing or updating Standard Operating Procedures (SOPs) in synergy with ongoing development efforts
• Mentoring the inexperienced development team from the inception of a compound right through to marketing approval equipping the R&D team with the appropriate knowledge and skill set
• Managing interactions with potential alliance partners (i.e. Big Pharma) is an option that is not usually offered by large CROs but can quite easily be performed by a consultancy as their experience is more comprehensive.
Innovation
From a scientific perspective, many of the tools used today to predict and evaluate product safety and efficacy are outdated. There has been a lack of investment in less common (unprofitable) diseases or risky innovative approaches and to apply new scientific knowledge in areas such as gene expression, analytic methods and bioinformatics to medical product development. There exists opportunities to create more effective tests and tools however, this will entail an initial investment of resources before these innovations are turned into reliable applied sciences. Although this may not yet be an option for developing biotechnology and pharmaceutical companies, they should still be considered for future consideration once revenue streams begin to flow.
Overcoming the challenges
Drug development is much more than designing a clinical trial. However as we have seen, this is by far the most lengthy and costly stage of the overall process. It is therefore imperative that developing biotechnology and pharmaceutical companies consider the causes and trends responsible for the burgeoning cost of pharmaceutical innovation before embarking on their own development programme.
Rising cost of drug development together with the continuing high clinical failure rate, increased regulation, competition and reimbursement efforts are on a collision course with societal demand for more cost effective accessible treatments.
A new development model or paradigm is needed, however this must delivered by Big Pharma as due to limited resources the only real options available to developing biotechnology and pharmaceutical companies are outsourcing alternatives.
For a developing biotechnology or pharmaceutical company, the prospect of embarking on the complex drug development process seems to be daunting and may even appear unaffordable. Employing the services of specialist niche service providers can help mitigate this hurdle.
Author Bio
Harjit Singh is a Clinical Research Professional currently based in London. He has international expertise in drug development with several global players and providing strategic planning and execution of global clinical development plans.





