
The Pharmaceutical industry has been growing in parallel to human evolution. Either inventing path breaking medicines for some of the incurable diseases or eradicating the viral threats from their roots; the industry has put in enormous efforts for the safety of humankind. In the last two decades, the industry has seen a new shift only to improve the efficiency and effectiveness of the medicinal production. In a way to be flexible and qualitative while benefitting the patients, the industry has been mainly focussing on bringing out the new classes of drugs and new-age technologies such as gene therapy, stem cells, nano-medicine, new drug delivery systems, etc. Standing with the industry’s growth, the global health authorities, too, endorsing the patients’ safety, have been coming up with new drug approval systems, compliance best practices, and procedures. In this scenario of 3600 evolution, how should companies design their plan of compliance? What should be their main focus is what we are going to cover in the following sections.
It is widely known that it takes an average of 14 years and 350 million dollars for a pharmaceutical company to get a new drug from a laboratory to the market. As each phase of these 14 years is completely regulated, companies should carefully plan the entire drug life cycle, right from the patent application, marketing approval, post-market sustenance and amendments to the patent expiration. In addition, they must consider phase-wise drug regulations to withstand global competition.
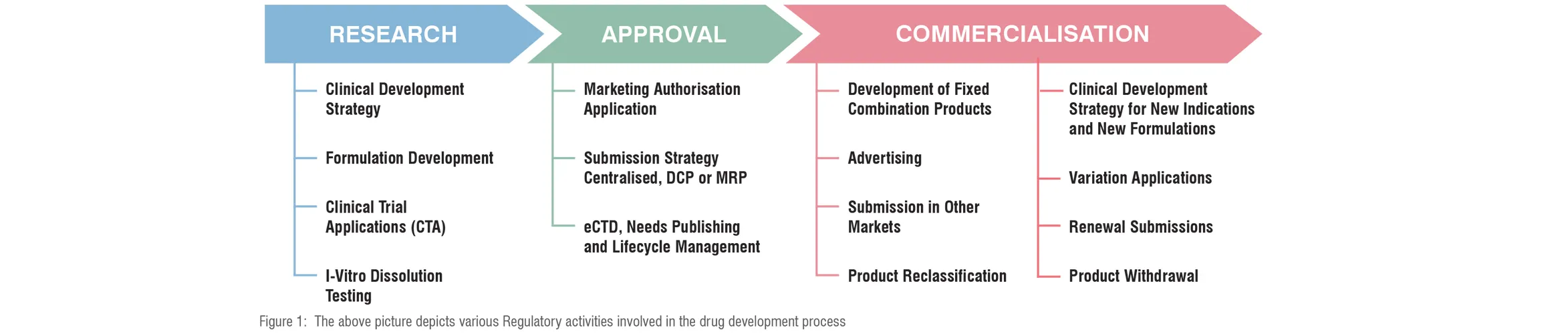
Phase-wise Regulatory Outlook
Health authorities control every stage of the drug development procedure including post-approval and the marketing stages as well. For the benefit of their assessment, evaluation, validation, and review procedures they mandate manufacturers to follow certain guidelines and best practices right from research to approval to commercialisation to post-market amendments as depicted below.
Let’s discuss these phase-wise regulations and procedures in detail.
Preclinical Testing
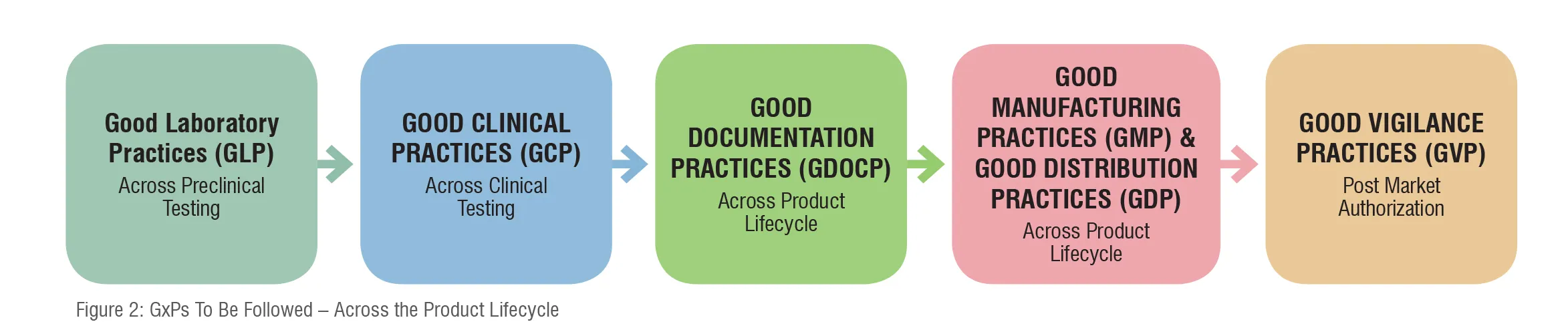
New drug development starts with the pre-clinical stage where pharmaceutical companies examine thousands of compounds to find a therapeutic value targeting a specific disease. The preclinical studies have to be conducted by following the Good Laboratory Practices (GLP) as suggested by respective health authorities. The principles of GLP govern the planning, performance, monitoring, recording, reporting, and archiving of preclinical studies. In this phase, the drug undergoes trials (in vitro, and in vivo) on laboratory animals to evaluate metabolism (pharmacodynamics [PD] and pharmacokinetics [PK]), safety, toxicity, dosage, and efficacy. Companies are obliged to show enough evidence to the respective health authority in the form of an Investigational New Drug (IND) application for the drug’s chemistry, manufacturing, and controls (CMC) to assure the identity, strength, quality, and purity of the drug.
Out of various compounds subjected to testing, if any of the drug compounds show promising results, only then can a company file an IND application. If safety concerns arise in the IND review, the health authorities can place the application on partial or full clinical holds. Often, such a case might be a disaster for companies striving to move forward to conduct their clinical trials.
Clinical Trials
The objective of clinical trials is to evaluate the safety and efficacy of medicinal product(s) in humans. Clinical trials involve four phases and each phase must comply with regional requirements as well as GCP as per the respective health authorities’ guidelines. The purpose of GCP is to ensure that all clinical trials adhere to ethical and scientific standards to protect the rights, safety, and well-being of trial participants as well as the reliability and credibility of trial results. GCP is also concerned with the data integrity.
Clinical trials held in the first three phases are conducted to collect safety and efficacy information to support the licensing application and phase IV is conducted post-marketing i.e., once the product reaches the market. The phase-wise details are as follows.
Phase I: Phase I clinical trials aim to find the best dose of a new drug with fewer side effects. These trials include initial single-dose studies, dose-escalation, and short-term repeated-dose studies. The goal of Phase I clinical trials is to determine what the drug’s most frequent side effects are and, often, how it is metabolised and excreted. If the drug is found to be safe enough, it is appropriate for further testing.
Phase II: Phase II clinical trials are considered as exploratory trials (as in this phase further assessment of drug safety is evaluated). Clinical pharmacology studies are also included in this category. Phase II trials begin only if Phase I trials don’t reveal unacceptable toxicity.
The drug, if found to show positive results is then considered for Phase III testing. At the end of phase II clinical trials, the manufacturer will have to discuss and update the health authorities regarding the development process, and the protocols for phase III clinical trials, which is the most extensive and expensive process of drug development.
Phase III: Phase III clinical trials or confirmatory trials are conducted to obtain additional information about the drug’s effectiveness and safety to assess the benefit versus risk of the drug. If approved by the health authority, the trail data can then be utilised for drug labelling process.
During the clinical trial stage, improper study design, cost computability, timeline, and lack of knowledge of pharmacovigilance may affect the company’s plan of compliance. Apart from these, companies must also review and check the accuracy of all submission materials in comparison with guidance documents from the Regulatory agency and safety topics from the International Conference on Harmonization (ICH).
Across the clinical trial life cycle, different types of data are generated, which has to be accurately reported to address specific research questions. Companies use this data to submit detailed Clinical Study Reports (CSRs), which forms the basis for marketing applications. Inaccurate or incomplete data may impact the product approvals. Therefore, maintaining data integrity is a mandatory requirement throughout the drug development process. Manufacturers are also obliged to follow Good Documentation Practice (GDocP), which applies to the creation, maintenance, and retention of documents.
Approvals
Once the phase III clinical trials are completed, and if the data demonstrate the safety and effectiveness of the drug, the manufacturer will have to compile the trial data to file a NDA or a Biologics License Application (BLA) depending on the type of the product. NDA or BLA usually comprises all the data, related to nonclinical and clinical testing and manufacturing, which must be in accordance with the Regulatory requirements.
In addition, while filing the NDA or BLA, manufacturer should consider the preferred regional format for submission either paper or electronic. Only once the Health authority approves the NDA or BLA, the manufacturer can commercialise the drug in their jurisdiction. If there are any deficiencies (major/minor) found in the data submitted, and the data provided with respect to safety, efficacy, CMC and labelling information is found to be inadequate, the health authority may reject the application and would ask for necessary clarifications.
Marketing and Commercialisation:
Once the drug is approved, manufacturers need to submit marketing authorisation applications in every country as per their targeted market-entry list by following GMP. Compliance with GMP is necessary in obtaining a marketing authorisation. This is because the health authorities validate the quality of drug products by carefully monitoring drug manufacturers’ compliance with GMP regulations. The GMP regulations govern manufacturing facilities, methods, processing, controls, and packaging of a drug product to ensure the identity, strength, purity, and quality of a product is appropriate to its intended use.
Apart from aligning to the GMP, companies must also follow the Good Distribution Practices (GDP) to ensure that there is no alteration to the drug’s innate formulation and to its package elements during the distribution.
In case if the drug is already approved in one country and the targeted country considers accepting the available marketing authorisation, the process can be a little easy for manufacturers. If it is not the case, the level of scrutiny would be higher. Then manufacturers may face challenges in terms of compiling qualitative region-specific clinical documentation, providing supplemental data during review stages if any deficiencies are figured out, and bridging evidence gaps, if any. En-route they may also have to consider targeted country’s assessment methods for marketing authorisation applications.
Pharmacovigilance(PV) and Quality Monitoring
Once the drug is approved and released into the market, manufacturers will have to conduct post-marketing surveillance to evaluate the performance of the drug or to report any unwanted and dangerous reactions. This is also known as Pharmacovigilance (PV).
Like other stages, PV too has its significance in controlling the drug’s market position. Though the manufacturer’s core responsibility lies in developing and releasing a safe drug into the market, they also need to track the drug’s performance and its effect on end users once it is released. If any adverse reactions take place, they should shoulder the responsibility of confronting it. Health authorities globally especially of the U.S. and Europe take adverse reactions seriously and mandate the industry to follow Good Vigilance Practices (GVP)to track the drugs’ performance once they are out in the market. The purpose of GVP is to ensure that continuous safety monitoring activities take place and all appropriate actions are taken to reduce the associated risks.
Hence, a manufacturer is expected to have a well-established PV system in place to monitor drugs’ safety cautiously and submit periodical reports about the benefit-risk profile of the drugs. In case of any discrepancy found in the post-marketing surveillance, there is a chance that health authorities may ask the manufacturer to withdraw the drug from the market. To avoid non-compliance and associated penalties, manufacturer must ensure that their PV systems are aligned to evolving regulations.

Though the phase-wise Regulatory outlook seems almost similar for many countries, there exist certain diversifications in obtaining approvals. Even if the procedures are different, the aim is mutual i.e. to protect the end user’s safety. To protect the same, the need of the hour is to harmonise the regulations, drug development, and approval procedures across the globe. Health Authorities should look upon a common global platform to govern and regulate the industry and bring in more centralised and commonly acceptable procedures.
This way, many innovative products can see the light of the day globally as soon as they are invented, and many unmet patient needs are addressed within the timelines. While Health Authorities should start working on centralised regulations, companies, with a unified view across development, approval, marketing and quality-monitoring stages, should build a robust plan of compliance i.e. to keep abreast of global standards and regulations and apply best compliance practices right from the first step (preclinical) to patent expiration.





