
The biopharmaceutical industry is currently experiencing a very challenging environment. There are many indicators: lower number of product approvals; consolidation among companies; fewer Big Pharma companies among the top 100 companies; decrease in the relative profitability of drug manufacturers; decline in the rate of increase of pharmaceutical sales; the variability of venture capital available to biotech; and, the increasing proportion of sales volume occupied by generic drugs in major markets.
How did this happen? According to some industry executives and analysts, production of new drugs is cyclical and the 2000s is merely a downswing after a very productive period throughout most of the 1990s. This slow down has caused a bubble in the flow of new products through the pipeline into the marketplace, exacerbated by increasing unpredictability of the longevity of products reaching the marketplace. Although a recent Tufts Center for the Study Drug Development (Tufts CSDD) study revealed a 52 per cent increase in the number of investigational compounds entering clinical development in 2003-2005 compared to the 1999-2002 period for the top 10 pharmaceutical companies, basic development obstacles such as time and cost of development as well as low success rates remain formidable challenges to the industry’s ROI. Another view is that there is a disconnect in the R&D continuum that occurs during what FDA terms the Critical Path, a period when compounds attain proof-of-concept and “go / no-go” decisions are made. In this view, the failure of the regulatory scheme and applied sciences to keep pace with the output from the discovery end of the R&D continuum have thus caused a productivity bottleneck in the development phase. Yet another perspective on what ails the clinical research enterprise calls the current system “fragmented, outdated and ailing,” and chronically vulnerable to funding uncertainty. Whether the problem is margins, markets, or myths, at one end of the pipeline or the other, the real question that confronts industry, policy makers, regulators, and stakeholders is how we progress from challenge to change.
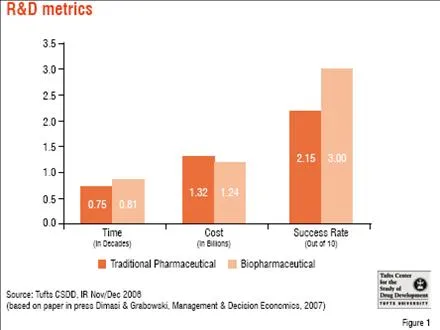
Industry metrics – What they tell us about how far, how fast The cost of development of a new pharmaceutical was estimated at US$ 802 million by a 2003 Tufts CSDD publication, based on a study of drugs with approvals from 1992 to 2001. This figure included the cost of failure as well as opportunity costs (i.e. what the investment would have garnered in the decade or so that it took to go from “lead to launch”). The study showed a 2.5-fold increase in the overall cost of drug development from the 1980s to the 1990s. An updated study on a more recent cohort (by approximately five years) of new approvals with broader representation of products from both biotech and pharmaceutical firms demonstrated that the overall cost had again risen, this time by 60 per cent, with less than expected differences in the costs of small versus large molecule development programmes.
Time is money
Another factor contributing to the escalation in the cost of drug development is that the time it takes to bring a drug through the clinical development. Average time taken for a regulatory approval process has not changed significantly in several decades and remains at about 7-8 years. While there were some gains in terms of clinical time during 1990s and 2000s, pre-clinical time increased considerably. One could attribute this to the 10-fold increase in potential drug targets due to the elucidation of the human genome, and the difficulties in validating these biological targets and identifying investigational compounds that affect them in useful ways. FDA review time has more or less stagnated after some dramatic improvements in the mid and late 1990s compared to the 1980s.

Success Rates – Obstacle to productivity
Success rates are another challenge that remains an obstacle to productivity. Success rates for new small molecule drugs hover between 10 per cent and 20 per cent, with an even wider range for large molecule drugs, depending on the therapeutic areas and types of compounds studied, the time frame, as well as the study methodology. The impact of the cost of failures has been estimated to be as high as 75 per cent of the cost of drug development. Failure rates for later phases in the drug development timeline are increasing, and have surged from around 30 per cent to 50 per cent in phase III. These late-stage failures are not only more costly in terms of time and money, but also in terms of resources diverted that could have been expended elsewhere.
This level of investment, in both absolute and relative terms, is considered “unsustainable” by regulators as well as industry executives and analysts. Modelling by Tufts CSDD indicates that success rates are the most attractive target in terms of impacting the overall cost of drug development. Increasing success rates by just 5 per cent and 10 per cent could reduce the cost of drug development by 14 per cent and 33 per cent, respectively. By comparison, reduction in pre-clinical costs would have to be on the order of 30-60 per cent, and decreases in clinical development time would have to reach 20-40 per cent to achieve the same effect.
The way forward – Process, products and paradigm
Process of development is in critical need of repairs. Recent work by the Tufts CSDD on the efficiency of innovation reviewed several dozen innovation initiatives worldwide (e.g. FDA’s Critical Path Initiative, the EU’s Innovative Medicines Initiative, the HapMap project, involving the UK, US, Japan, China, Canada and Nigeria).
The clinical phase is the lynchpin of the R&D process. Much attention has been devoted recently on how to break bottlenecks at this phase. Biomarkers are key to assessing safety and efficacy of investigational compounds early in development to decide which ones should proceed to later stage trials, and confirming the utility of drugs in pivotal trials by predicting the ultimate clinical benefit of a drug without having to wait for long-term outcomes such as survival time. FDA has initiated a programme of Voluntary Exploratory Data Submission (VXDS) together with the European Medicines Agency (EMEA) in order to expand the science base, instill public confidence and improve regulatory decision-making on pharmacogenomic data and its uses. Adaptive clinical trials and EDC are methodological and operational processes that are expected to improve the efficiency of clinical trials. Microdosing is one of several exploratory IND approaches that should promote earlier “go / no-go” decisions, reducing the number of compounds brought forward into the clinic as well as better characterising the ones that are targeted for further testing. This could reduce costs, but the initial impact is likely to be in the form of increased success rates.
How fast the technological fix for the clinical phase is progressing can be seen in Figure 2 based on recent surveys of utilisation of new approaches in trials currently in progress. However, anecdotally drug developers have reported that rates for trials just getting underway approach 90 per cent for incorporating EDC and pharmacogenomic testing. Meanwhile, on the regulatory side, FDA is emphasising its openness to adaptive designs, has also published a list of genomic biomarkers that have been used successfully to support approval, and together with the EMEA and MHLW, is working on a proposal for harmonising the qualification of pharmacogenomic biomarkers.
While improved process will address some of the challenges, others are related to the products themselves. The FDA points out that even a commonly used drug, such as warfarin, has been found to have a 7-fold inter-individual variability for dose. For this reason, the FDA wishes to move from a population-based model of product development in which one-size-fits-all, to a more targeted approach based on pharmacogenomics, i.e. personalised medicines. While these medicines will not be individually tailored per se, they will be increasingly limited to smaller subpopulations of patients evidencing certain genetically identifiable responses to drugs. This will increase the current momentum within the industry to move away from the highly competitive primary care market model based on blockbusters to speciality care markets (typically characterised as being biotech-derived, requiring special conditions of administration, and having high-cost-per-treatment cycle).
Identifying and developing plans to manage product safety prophylactically through risk management plans may increase the cost of drug development in the short run, but in the long run will hopefully decrease time of development, while laying the groundwork for a return to confidence among practitioners, patients and investors. Some regulatory process initiatives, however, have proven successful already. For example, under the FDA’s fast track programme, products intended to treat serious and life-threatening conditions with unmet medical needs are given special attention by the FDA through intensive scientific interaction. Other such programmes target products for rare diseases or illnesses for which the benefit-risk balance indicates that the need to make treatments available surmounts the regulatory requirement for completing the dossier until after approval (i.e. accelerated approval). Japan and the EU have similar programmes. Review of FDA approvals for the last decade indicates that the number of new approvals for such special populations has increased from 20 to 40 per cent, yet the subsequent safety history of these products in terms of safety withdrawals has been better than new approvals overall.
This changeover in product focus could serve three major goals: increasing industry’s economic health by decreasing its reliance on blockbusters; increasing its attention towards emerging markets; and, better serving public health by directing more R&D resources towards unmet medical needs. There are signals that this is indeed happening. A 2007 IMS report indicated that emerging markets contributed 27 per cent of the growth in world sales, double the rate in 2001. Also, of the 105 global blockbusters, almost half were specialist-driven, up from just 14 in 2000. Yet, there is much room for growth in these emerging and specialised markets. As of 2002, WHO estimates that there are some 2,000 diseases worldwide considered to have unmet medical needs.
While changes in the process and product realms will help address some of the challenges facing the industry, the paradigm itself will have to evolve before sustainability and profitability become the mainstays of a healthier and health-creating biopharmaceutical industry. Regardless of which way biotech is going, it is difficult to separate the fate of biotech from Big Pharma. Wealth Daily reports that in 2007 nearly US$ 100 billion was spent in Europe and the US on biotech mergers and acquisitions, much of it by Big Pharma, which may be strapped for products, but not cash. While Big Pharma is absorbing biotech products, companies, and platforms at an increasing rate, and shape-shifting into a new industry, it will also have to economise to sustain itself. This will demand rapid reapportionment of R&D resources from highly-competitive therapeutic areas in primary care to more specialised therapeutic areas, utilising process redesign to shorten clinical development time, decreasing costs and increasing patient recruitment by outsourcing clinical trials to sites in emerging economies, and offshoring of ancillary pharma business segments.

Challenge to change
In response to evolutionary stress, the R&D paradigm itself is undergoing metamorphosis, albeit a circular one. The biotech revolution was made possible by a revolutionary business model, spin-offs from academia funded by venture capitalists. The scientists that formed the core of the movement were highly innovative, but also highly specialised, and thus vulnerable to economic extinction events. This left the door open for Big Pharma to survive, both competitively through horizontal integration and adaptively through vertical integration. The tableau has come full circle. Now markets are becoming specialised with the prospect of further movement in this direction under pressure from personalised medicine. The R&D niches of the public and private sectors increasingly intersect. Biotech spin-offs often rotate back within the sphere of Big Pharma or coalesce into big biotechs. This has given rise to the emergence of an all-encompassing biopharmaceutical sector highly charged to innovate technologically, organisationally, and commercially. Yet in order to thrive, it must first survive the rough road from challenge to change.
References
1. Kaitin K., editor. New drugs entering clinical testing in top 10 firms jumped 52% in 2003-05. Tufts Center for the Study of Drug Development Impact Report May/June 2006; 8(3)
2. Crowley WF et al. Clinical Research in the United States at a Crossroads. JAMA 3 Mar 2004; 291(9):1120-26
3. DiMasi JA et al. The price of innovation: new estimates of drug development costs. Journal of Health Economics 22 (2003) 151-185
4. DiMasi JA, Grabowski HG. The cost of biopharmaceutical R&D: is biotech different? In press: Managerial and Decision Economics
5. Pharmaceutical Executive January 2006:54
6. DiMasi JA. The value of improving the productivity of the drug development process: faster times and better decisions. PharmacoEconomics 2002; 20 Suppl. 3:1-10
7. Frueh F, Associate Director for Genomics, Office of Clinical Pharmacology and Biopharmaceutics, CDER, FDA. PGx and biomarkers: Current role in drug development. Accelerating Anticancer Agent Development and Validation, Bethesda, Maryland [Slide Presentation] 2 June 2005. Attributed to: Hillman MA et al. Relative impact of covariates in prescribing warfarin according to CYP2C9 genotype.
8. Engel S. How to manage a winning pipeline. R&D Directions 2002 Feb;8(2):36-41
9. Xanthopoulos KG. Producing more and Better Drugs. San Diego Union Tribune 19 January 2006 available at http://www.signonsandiego.com/uniontrib/20060119/news_lz1e19xanthop.html, last accessed October 17, 2006
10. Increased costs of product deals requires change of strategy, says Wood Mackenzie. Pharma Marketletter 27 March 2006: 25
Read the full article — it's free
Register with Pharma Focus Asia to unlock expert insights, research articles, and in-depth industry analysis.





