When a compound progresses to a medicinal product the complexities of disease, drug action, and the diversity of patient response can mean that nuances of drug behaviour may not be fully evident. However, new insights may emerge on more widespread use. These can provide ideas for new indications or better treatment of the existing indication.
Programs to identify and evaluate novel therapeutic agents are expensive, take a long time and do not guarantee a successful outcome. Proving safety and efficacy is difficult, complex and slow. Furthermore, even after efficacy and safety are demonstrated and the compound becomes a medicinal product the complexities of disease and of the drug action, allied with the diversity of patient response can mean that nuances of the new drug’s behaviour may not be fully evident. Consequently, most new medications are not perfect. Recent restrictions and withdrawls of the Cox-2 Inhibitor drugs for arthritis exemplify how difficult it is to determine each and every facet of drug performance in clinical studies. Much greater patient experience may be required.
As a medication becomes widely used, new insights emerge on modes of action, side effects and patient response. At the same time, advances in molecular biology, receptor pharmacology and better understanding of the clinical condition may create possibilities for new indications or for better treatment of the existing indication by altering dosage regimen or by dosage form redesign. Concurrently, technologies may emerge to enable more efficient, effective or convenient ways of delivering or even targeting the medication.
Possibilities exist, therefore, to redesign established drugs—usually by reformulation— so they are more effective, safer or more convenient for the patient.
The last decade or so has seen an explosive growth in Information Technology (IT), making data and information capture, analysis and dissemination widely available. More recently there have been moves to make detailed clinical trials data available in readily accessible databases (1). Such knowledge, allied to creative interpretation and linkage to other emerging facets of knowledge can be a rich source of opportunities for product redesign.
Information sourced in such ways may concern:
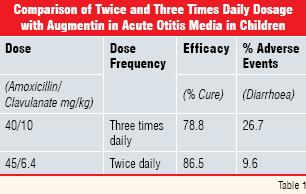
Development programs exploring safety and efficacy cannot possibly cover each and every permutation and combination of dose frequency and timing. Information from widespread use may suggest other more suitable regimens. The antibacterial amoxicillinclavulanate (Augmentin®) was administered three times daily when first commercialised, reflecting the rapid elimination rates of both the amoxicillin and clavulanate components. It proved to be a very effective therapy in community-acquired infections but three times daily dosage is less convenient (and may lead to reduced dosing compliance) than less frequent administration. Furthermore, diarrhoea was a frequent side effect, especially in children under two years of age. This was attributable to the clavulanate component.
In vitro evidence that lower doses of clavulanate coupled with a shorter treatment period might be equally effective, prompted the development of formulations for twice daily dosage. Clinical trials in paediatrics indicated that the twice daily presentation was equally effective to that dosed three times and that the diarrhoea side effect was significantly less as illustrated in Table 1 (data abstracted from Reference 2).
More detailed knowledge on metabolism, pharmacokinetics of the metabolites and possible impact on clinical behaviour is also likely to accumulate on widespread usage of the medicine. This may suggest that the dosage form could be redesigned. An example concerns Niacin, utilised as a lipid lowering agent. Side effects (flushing and hepatotoxicity) are related to its metabolic pathways. Metabolism proceeds via two pathways viz. conjugation and amidation. If the amount ingested and absorbed saturates the amidation route (as would be expected from a rapidly releasing dosage form), the conjugation route dominates, resulting in a higher incidence of flushing. Slower absorption can result in enhanced amidation and a relatively higher level of the metabolites associated with hepatotoxicity (Table 2).
Thus, dissolution rate from the dosage form can influence the metabolic profile and associated side effects. This knowledge allowed a formulation to be developed with drug release rate providing an appropriate metabolic balance to minimise side effects (3).
Drug-related improvements may also be stimulated by advances in separation sciences or purification techniques to provide a medication that may be more potent, has greater specificity or evinces fewer side effects. The Proton Pump Inhibitor, omeprazole, used to treat gastric ulceration and related conditions is a racemic mixture. The single isomer esomeprazole has now largely replaced it because of better ulcer healing rates.
Purity-related improvements have also been recorded for biopharm products. Recombinant human insulin reputedly has less side effects than porcine-derived material. Similar improvements have been recorded for recombinant human growth hormone. It has also been reported that a “new formulation” of ß interferon-1a is better tolerated and induces less antibody formation when treating multiple sclerosis (4).
Molecular biology
Knowledge of the aetiology of diseases is continually expanding. Two decades ago there was no evidence that gastric ulcera tion is caused by bacteria in the gastric mucosa. In 2005, the scientists who identified and elucidated the role of Helicobacter in gastric ulcers were awarded the Nobel Prize for Medicine. Aspirin was used as an anti inflammatory and analgesic for almost a century, before its prostaglandin inhibition mechanism was elucidated and its beneficial effects at low dose as a platelet aggregation inhibitor established. Such findings have led to the use of antibiotic preparations, possibly in conjunction with proton pump inhibitors or other anti acid agents for treating gastric ulcers and of low dose aspirin as an anti-clotting agent.

Increased insight of molecular biology, whether of the disease or mechanism of drug action may also suggest a drug’s potential in related or even different clinical conditions. In such a context, the anti arthritic drug celcoxib (Celebrex) is currently being evaluated for efficacy in colon cancer (5).
Time-related effects
Symptoms in some disease states can be manifested at different times of the day, or even seasons of the year. Seasonal effects can be environmental (e.g. Hay Fever or other allergic conditions are more acute at the height of the pollen season) but others can be more complex. Time-associated effects include:
• Asthma: Airways obstruction can be most severe early in the mornings
• Rheumatoid Arthritis: Joint stiffness is usually most severe in the morning. Conversely, pain from osteo-arthritis is more prominent in the evening
• Cardiovascular incidents: Heart attacks occur frequently in the early morning. This may be related to the stresses to the cardiovascular system, caused by going from a prone to the upright position after waking up. It is important, therefore, that beta blockade or other cardioprotective effects are optimal at this crucial time
• Hypertension : Blood pressure in normotensive subjects usually falls during sleep (“cardiovascular system rests”). However, in some hypertensives this does not happen. The increased incidence of nocturnal heart attacks at around 2 AM to 3 AM is ascribed to the cardiovascular system remaining under stress
Awareness of such circadian effects can stimulate ideas for timing drug delivery to coincide with the chronotherapeutic requirements of the clinical condition. The timing of the cardiovascular events listed above has led to the development of a medication of the calcium antagonist, verapamil that delays delivery of drug (the medicine being taken at bed time) until about 8 hours after dosing to cover the vulnerable early morning period (6). The drug is thereby delivered to the receptor at the time of greatest need.
Other examples of delivering drug “when required” concern theophylline and salbutamol. These drugs have relatively short half lives with concomitantly short durations of action. Formulation as delayed/sustained release formulations are calculated to provide adequate plasma levels to cover the vulnerable early-morning period. Sustained release theophylline also reduces its peak plasma level, thereby mitigating the effect of its low therapeutic index.
Seasonal effects, other than environmental or climatic ones are lesser known but endorphin levels are apparently greater during January/February and lowest in July/August. One could speculate as to whether this might mean that cancer therapy would be optimal at times when the patient might be more resilient to aggressive treatment. Could vaccination be more effective if aligned with season-related levels of natural mediators? There is no evidence for such effects but such relationships might merit exploration.
Infectious diseases
Bacterial resistance to antibiotics is not new; it has always been a race between resistant organisms evolving better defence mechanisms and scientists finding novel antibacterials. However, few novel antibacterials have emerged in recent times and resistance by pathogenic bacteria is a major issue. The prevalence of methicillin-resistant S.aureus has been well-documented. Drug resistance by pseudomonas aeruginosa is also increasing. The problem is not confined to hospitals or Intensive Care Units. Resistance in community-based infections is also a concern. Penicillinresistant Streptomyces pneumonia are now prevalent in many countries. In the light of all these considerations it is necessary to consider new approaches to treating infections using existing drugs.
New treatment paradigms
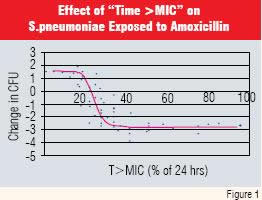
The emergence of bacterial resistance caused Craig W.A. and Andes.D to develop the “time above MIC” concept for antibacterial therapy. The principle is that if levels of some antibiotics are maintained above the minimum inhibitory concentration for a specific time (“T>MIC”), efficacy is enhanced (7). Woodnut et al, working with resistant strains of Streptomyces pneumoniae confirmed this thesis for the broad spectrum antibiotic amoxicillin. They showed that the minimum “T>MIC” for amoxicillin was about 35% of the dosage interval (Figure 1 and Reference 8).
Thus, if amoxicillin is administered twice daily, and serum levels exceed the inhibitory concentration for a minimum of 4.2 hours (35% of 12 hours) it is likely to be effective against penicillin-resistant Streptomyces pneumonia. This finding provided an opportunity for redesigning amoxicillin-containing dosage forms as findings in animal studies can be more readily translated to clinical efficacy in patients as the bacterium can be considered “the receptor” in both instances.
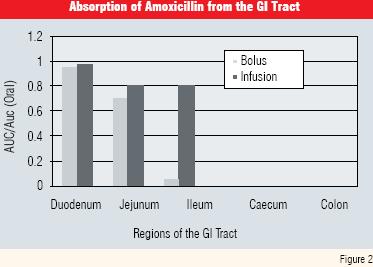
However, the rapid clearance rate of amoxicillin (half life of about 1 hour) militates against persistence in the biosystem. This, together with its limited “absorption window”, makes it difficult to formulate as a delayed or prolonged release formulation. This is because, although it is absorbed very well in the upper regions of the duodenum absorption becomes progressively less efficient in the jejunum and ileum and is almost non-existent in the colon (Figure 2 and Reference 9).
Furthermore, “unabsorbed drug” could kill symbiotic bacteria in the colon, causing gastro-intestinal side effects because of overgrowth of resistant bacteria.
The formulation developed to provide sustained plasma levels utilised a novel approach, incorporating two salts of amoxicillin viz. the conventional trihydrate for “immediate release” and the sodium salt, co-formulated with citric acid as a release modifier to deliver a later dose. The plasma level profile is shown in Figure 3, with a “time above MIC” (4mcg/ml) that is almost 50% of the twice daily dosage interval (10).
This formulation was shown to be effective in Phase 3 clinical trials and is now a commercial product in the US and some European countries.
Prolonging drug in the biosystem for the requisite time to optimise bacterial kill rate is one example of changed paradigms providing more effective therapy. More recently, the concept of dosing “pulses” of drug that provide limited time and duration in the biosystem has also been propounded (11). The concept has been validated in vitro with antibiotics and antifungal agents but is also being suggested for other clinical conditions including antifungal and cancer therapy.
Not every drug will be suited to pulse dosing. Drugs with long half-lives will not be cleared quickly enough to get true pulse profiles. It is also likely to be difficult to deliver pulses orally; the gastro intestinal tract is too variable to facilitate such precise plasma profiling. Pulse delivery is probably best suited to parenteral administration. Variables such as the frequency and amplitude of the pulses may be critical and need to be explored and defined. It might well be that different organisms or clinical conditions require different pulse profiles. Programmable pump systems could be utilised to reliably deliver the requisite pulse profile.
Knee/hip joint replacement techniques have revolutionised the treatment of joint inflammatory disease. Cardiac stents have had a similar impact in coronary bypass surgery. But they have also brought new challenges. Bacterial infections, localised at the replacement site are notoriously difficult to treat and frequently require further invasive surgery. Consequently, a need and associated opportunities exist for anti-infectives, embedded in the joint or stent material to be released slowly to evince a local antibacterial effect.
Chronic low dose therapy is also being considered for treating cancer. Traditional chemotherapy usually involves giving the patient the highest dose they can tolerate to maximise tumor reduction. But such drugs are toxic, so dosing must be periodically halted for several weeks to allow the body to recover. It has been suggested that such “rest periods” can allow a tumor to rebuild the blood vessel network it needs to keep growing. So, researchers are experimenting with giving very low doses of cytotoxic drugs for extensive periods of time. Patients are spared the toxic side effects associated with high dosage, so treatment could be continuous and long term. This could help turn some cancers into chronic, manageable conditions. Promising results are being reported from clinical trials on cancers like brain tumors, non-Hodgkin’s lymphoma and breast and ovarian cancer (12).
Prolonged administration of low doses will require more convenient, non-invasive modes of dosing than are now utilised with cytotoxic agents, again providing opportunities for developing new formulations or delivery systems.
Demographics
Better diet, living conditions and healthcare programs mean that people are now living longer in many regions of the world. Consequently, diseases of old age have become more prominent. For instance, the incidence of Benign Prostatic Hyperplasia (BPH) in males at age 60 is about 50%. By age 70 it rises to 70%-80% and by age 85 incidence is in the region of 85%.
Many of the elderly remain active, alert and independent because of their lifestyle, sensible dieting, and diligent use of medication, or, in many cases, several medications. Convenience can greatly affect compliance in chronic dosing scenarios. For instance, if all the medications could be incorporated in a single unit it would enhance convenience, compliance and consequently effectiveness. If such “multi-drug” medications could be designed to deliver each drug at the right time and possibly the appropriate rate to reflect differences in clearance rates, or in other metabolic processes in the elderly, it could be enormously beneficial. A drug for insomnia might be designed to take effect quickly whereas a medication for emphysema might be released later from the dosage form. In addition to such chronotherapy-related delivery, a multi-medication unit might be designed to gradually release drug to avoid saturation of biosystems enzymes or pharmacological interactions. There are immense opportunities in geriatric medicine to provide more user-friendly and more effective medications.
Pharmacogenetics and pharmacogenomics
The DNA sequence of the human genome (the “physical map”) has been elucidated. The functional aspects are also being progressively mapped and a stage may be reached where it is possible to predict many biological responses and susceptibility to particular diseases or enzyme deficiencies. Evidence is being accumulated that drug response may be influenced by individual genomic networks. This may provide greater insight on what medication might or might not be appropriate for specific individuals, leading to more focussed, more effective and safer pharmacotherapy. It might also stimulate demand for “personalised” medications, requiring greater flexibility in terms of dosage, possible rate of drug input (release rate from the dosage form) than is now thought necessary. Systems and technologies need to be developed to provide such flexibility but, when demand emerges, technologies, ideas and associated opportunities invariably follow.
Paediatric medications
Medications are evaluated primarily in adults in the first instance because of ethical, regulatory and “informed consent” considerations. Traditionally, paediatric medicine has usually been an “add-on” to adult therapy. It is now accepted that the premise that plasma levels and kinetics established in adults evince the same effects (on a mg/kg basis) in paediatrics can be flawed. Children can differ from adults in metabolic and clearance capability (and such characteristics change more rapidly over time than with adults). Scaling down the dose, based on body mass considerations may be too simplistic. Paediatric-specific medications may be necessary.
Patient convenience
Adult dosage forms (usually a tablet or capsule) are often unsuitable for dosage to young children because of size and swallowing difficulties. A liquid product is preferable but palatability then becomes critical. Taste is usually a function of the level of drug in solution. If concentration in the dissolved state exceeds the bitterness threshold there is little that can be done by simple formulation to improve matters. Sweeteners like sucrose can elevate the bitterness threshold to some extent but sucrose is now considered undesirable in medicinal products. Other techniques to mask poor taste (e.g. coating with polymeric materials) are generally of limited value. Bioavailability may be compromised and in any case coated particles (because they are likely to be larger than drug particles) can have a gritty texture and may be broken by mastication, with the drug being “released” in the mouth and evincing the bitter taste.
More effective ways must, therefore, be used to improve palatability of liquid products. One approach is to utilise a form of the drug (e.g. different salt form), whose solubility is lower than the bitterness threshold. However, depression of solubility can affect absorption rate and bi oavailability. In very general terms it is accepted that, when solubility exceeds about 1-2mg/ml absorption may not be an issue (13, 14). This suggests that, where bitterness threshold is in this region, the use of an alternative salt offers possibilities for taste masking with no impact on bioavailability.
Drug-device combinations
Many useful systems have been developed to facilitate parenteral administration (particularly self-injection), diagnostic testing (e.g. blood glucose monitors) and for inhalation dosage. The search for a non-invasive way of delivering insulin has lasted a long time. Indeed, many despaired that there would ever be a successful outcome. The major barriers concerned insulin’s peptide structure, making oral delivery unreliable, if not impossible. The steep dose-response relationship and need for dose to reflect blood glucose levels were also considerable constraints. Persistence has paid off, however, and a system for insulin delivery by inhalation has now been tested in clinical trials and shown to be safe and effective. The product shown in Figure 4 is now commercially available.
Time will tell whether inhaled insulin becomes widely used and whether such delivery is appropriate for patients suffering from other conditions that cause breathing problems (emphysema and asthma). At the same time, other devices for inhalation delivery of insulin are being developed. It is the nature of such competition that each new product represents an incremental improvement over existing ones. Over time, such cumulative enhancements lead to significant improvements.
Incremental improvements have also featured prominently in inhalation products for treating asthma. The first systems for delivery of dry powder were effective but required manipulation of drug (which was contained in a capsule) and device prior to dosage. In young children, this could be embarrassing for dosage while at school and on social and sporting occasions. Devices are now available that do not require cumbersome pre-dosing assembly so are more convenient and acceptable to the patient (Figure 5).
Drug-device combinations are likely to continue to offer great opportunities for innovative ways of drug delivery.
Novel diagnostic techniques are also likely to attain greater prominence in all patient groups. The Prostate-Specific Antigen test for early diagnosis of prostate cancer is now routine but there is great hope that similar diagnostic tests for autoimmune conditions, cancer, osteoarthritis and liver disease will become available over the next decade. It is conceivable that treatment paradigms for “early disease states”, identified by such techniques may differ from those applied to well-established disease states. This too should lead to existing drugs finding new or slightly different uses that make them better medications.
src="https://industry.pharmafocusasia.com/images/manufacturing/mmp_patrickcrowley5.jpg" />
Medicinal agents are structurally designed for optimum specificity and “potency” in most cases. They can be highly lipophilic and poorly soluble as a consequence. Yet, a material must dissolve in aqueous media to be transported to the systemic circulation (while being sufficiently lipophilic to partition from the aqueous intestinal contents across the enterocytes lining the small intestine). It may be difficult to balance such competing features in a chemical structure. Consequently absorption may be sub-optimal, variable or dose-constrained, leading to plasma level variations causing unreliable therapeutic response or greater incidence of side effects. Local side effects are also possible, consequent to poor absorption. Gastrointestinal side effects during treatment with broad spectrum antibiotics have been ascribed to unabsorbed antibiotic killing symbiotic bacteria and subsequent overgrowth of resistant flora.
Figure 4
Reproduced with Permision from Pfizer Inc.
Oral absorption enhancement has the potential to make the medication more reliable, by reducing the variability in plasma levels. Other potential advantages, if absorption enhancement were to be significant might be a lower dose, smaller dosage units and lower cost of goods for the medication.

Figure 5
Optimising absorption may involve using a different solid state form of the drug, altering the physical properties of the existing form, formulation with materials that enhance absorption, exploiting the physiology of the gastrointestinal (GI) tract or capitalising on pre-hepatic metabolic and transport systems. There is a wealth of literature to provide guidance on improving absorption. Hence, the topic is not reviewed in this article. Reference 15 reviews physico-chemical approaches to such enhancement. <
Performance enhancement of medications is invariably driven by new biological insights that may relate to the patient, the disease condition or the drug. Generally, evidence from clinical trials is required to validate and support any claims for improvement. However, overall there is less risk of failure with such programs than with novel molecular entities. The compounds being evaluated for improvements will have been thoroughly evaluated for safety, so the risk of attrition because of toxic effects is low or maybe non-existent.
Better understanding of how drugs work and increased access to such knowledge makes it likely that such opportunities will continue to become available for providing better medicines so that patients receive improved treatments to make them feel better and live longer.
1. FDAMA 113; http://prsinfo.clinicaltrials.gov/fdama-113.htm
2.Hoberman.A, Alejandro.M.D, Paradise.J.L, Burch.D.J, Valinski.W, Hedrik.J.A, Aronovitz.G.H, Drehobl.M.A, Rogers.M.J; Equivalent Efficacy and Reduced Occurrence of Diarrhea from a New Formulation of Amoxicillin-Clavulanate Potassium for Treatment of Acute Otitis Media in Children. The Pediatric Infectious Diseases Journal (1997) 16 (5) p 463-470.
3. Pieper.J.A; Overview of Niacin Formulations: Differences in Pharmacokinetics, Efficacy and Safety: Am.J.Health-Syst Pharm (2003) 60 Suppl 2; S9-S14.
4.Anon; PR Newswire (U.S.) 28 Sep 2006).
5.Anon: Wall Street Journal 19 Sep 2006)
6 Neutel.J.M, Alderman.M, Anders.R.J, Weber.M.A. Novel Delivery System for Verapamil Designed to Achieve Maximal Blood Pressure Control during Early Morning; Am Heart J (1996) 132 1202-1206.
7.Andes.D, Craig.W.A. In vivo activities of amoxicillin and amoxicillin-clavulanate against streptococcus pneumonia: application to breakpoint determinations. Antimicrobial Agents and Chemotherapy (1998) 42 (9) 2375-2379.
8.Woodnut G, Berry V. Pharmacodynamic models for assessing the efficacy of amoxicillin-clavulanate against experimental respiratory tract infections caused by strains of Streptococcus.pneumoniae. Antimicrobial Agents and Chemotherapy (1999) 43 (1) 29-34.
9.Barr.W.H et al; Clin Pharmacol & Therapeutics (1994) 56 (3), 279-285.
10.Kaye.C.M, Allen.A, Perry.S, McDonagh.M, Davy.M, Storm.K, Bird.N, Dewit.O; The Clinical Pharmacokinetics of a New Pharmacokinetically Enhanced Formulation of Amoxicillin/Clavulanate; Clin.Ther (2001) 23, 578-584.
11.Ibrahim.K.H, Gunderson.B.W, Hermsen.E.D, Hovede.Laurie.L.B, Rotschafer.J.C; Pharmacodynamics of Pulse Dosing versus Standard Dosing: In Vitro Metronidazole Activity against B.fragilis and B.thetaiotaomicron; Antimicrobial Agents Chemother (2004) 48 (11), 4195–4199.
12. Anon: Wall Street Journal 25 Oct 2005.
13.Florence.A.T, Attwood.D, Physicochemical Principles of Pharmacy; Macmillan, Third Edition, London (1998) p 21.
14. Hamdou.H.M; Dissolution, Bioavailability and Bioequivalence; Merck Publishing Co; Easton PA (1989) p368).
15.Crowley.P.J, Martini.L.G;. Physicochemical Approaches to Enhancing Drug Absorption; Pharmaceutical Technologies Europe (2004) 16 (9) 18-27.
-- Issue 20 --

Patrick J Crowley, Vice President Pharmaceutical Development GlaxoSmithKline USA.

Luigi G Martini, Director Process Technologies.













