Between 1997 and 2000 Pharmadule built a number of pharmaceutical manufacturing facilities in China for both domestic manufacturers and multi-national pharmaceutical companies, including Eli Lilly and AstraZeneca. The multi-national companies required GMP compliance and validation services comparable to the level that exist today. The facilities delivered for Chinese manufacturers would comply with international GMP requirements; but it became evident at that time that the Chinese GMP regulation was immature in comparison with the EU regulations and guidelines. In 2011, all of this changed when China launched the new GMP regulations which elevated the requirements to a level equivalent with international cGMPs.
At about the same time, Pharmadule refocused the strategy towards the Chinese market and has since then been carrying out Validation, Quality Management Systems, GMP compliance improvement, and design projects in China. Compared to the projects in the late 1990s, assignments recently undertaken required Pharmadule to consider a number of significant changes in the Chinese marketplace when designing a modernised approach for project execution in China and other emerging markets.
One major difference is that while European and American pharma manufacturers typically have a QA/QC-force amounting to up to 40 per cent of the production staff, the Chinese companies we have encountered have very small QA/QC-departments, mainly focusing on QC and batch release testing. Quality is typically tested and validated into the products and processes. Interestingly enough, these immature QA and QC practices partially enable the transition to modern validation planning and execution.
The reason is that, in contrast to the multi-national pharmaceutical companies Pharmadule has worked with, over the last 25 years, Chinese companies do not have bureaucratic and over-compliant Quality Management Systems and validation frameworks. Rather, they could be characterised as non-compliant with EU or US regulations. This is of course a concern when it comes to the current level of Quality Assurance expertise. However, our Chinese clients have been very open to embracing new ideas and knowledge, allowing them to evolve faster than any other market where Pharmadule has worked in the past. Organisations offer little resistance to change, provided they understand the benefits and the details of the new approaches. Consequently, implementation of new business processes can be much quicker than in Europe and the US.
When a new facility is being built, it provides the manufacturer an opportunity to completely change the approach to Quality Assurance and validation. Changes that would take half a decade or more to fully implement in a multi-national pharmaceutical company in the EU or the US are accomplished in a matter of months.
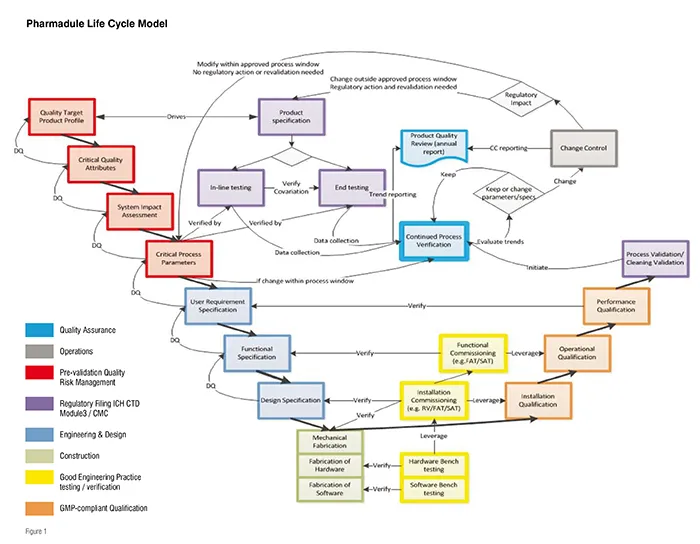
In recent assignments completed by Pharmadule in the Chinese market, the philosophy described by international regulators in ICH Q8 – Pharmaceutical Development, Q9 – Quality Risk Management and Q10 – Pharmaceutical Quality System have been seamlessly integrated with the concepts of Process Validation as described in the FDA Process Validation Guidance from 2010 and ASTM E2500 - Standard Guide for Specification, Design, and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment. Other international standards have also been taken into consideration in order to create a state-of-the-art Validation Master Plan. This Validation Master Plan governs the entire product and process life cycle (as shown in Figure1) and is used to manage all of the phases of an investment project. The main steps of this plan combine to form a control strategy allowing an unbroken chain of traceable verifications all the way from the product attributes (e.g., shelf life) via Quality by Design activities in the Process Design and scale-up, through design and qualification, all the way to process validation. This level of traceability is rare even with established international companies.
Implementing a new QA culture and a new paradigm for process validation has of course not been an altogether easy task. There have been a number of obstacles, some obvious, some less so. But there have also been circumstances in China that enabled the shift.
Key observations
It was a positive surprise to find that the degree of process understanding was very high among engineers, comparable or even exceeding western expectations. This can probably be attributed to the effectiveness of the Chinese education system. However, this process understanding was rarely fully leveraged into the process design and the GMP documentation. With the right tools and training, process understanding could easily be transcribed into Critical Quality Attributes, Critical Process Parameters and ultimately risk-based control strategies. Defining these boundaries and limits for the process in Quality by Design work has been both efficient and accurate. Quality by Design also facilitated, and eliminated unnecessary aspects of Technology Transfer, since Process Design departments, Operations and QA worked closely together. An impediment that could slow down changes is that within the Chinese culture, there traditionally is no challenge to authority. This puts a cap on the capability of innovative thinking and creative solutions. Corporate culture in China rarely encourages coworkers to take risks and explore new solutions. In fact, many companies punish employees that take risks and fail, with public shaming and fines. It is, therefore, important to note that, in contrast to the engineers, operators in the facilities do not have the same level of training as their western counterparts and will not take own initiatives. They normally only speak Chinese and will, for the reasons stated above, follow the SOPs they are given very rigorously. When training operators, this must be taken into account.
Turning process understanding and Quality by Design into User Requirement Specifications is a challenge that is not unique to Chinese manufacturers. International companies also regularly fail in this area. The sheer number of process engineers, support from management, detailed instructions and a flexible approach to change, allowed rewriting of the URSs to enable traceability of critical parameters and aspects. Revamping the URSs has consequently been easier than expected. This is a key activity in providing the foundation both for procurement and for the rest of the validation activities (In the new draft Annex 15 to the EU GMP guidelines, URSs are specifically highlighted as part of the validation process). In China there is an additional challenge that increases the importance of URSs.
Traditional Chinese vendor management pushes all the responsibility for design, commissioning and qualification — up until at least Operational Qualification — on the equipment vendors. Audits and contract management have not been common practices. On the contrary, maintaining good relationships between business parties is considered so important that relationships sometimes supersede written agreements. If the supplier is to play a big part in the qualification, as is the intention (such as in ASTM E2500) then the supplier must initially undergo an audit to ensure that they are able to provide risk-based documentation, next get a URS that allows them to write risk-based documentation and finally communicate during the design phase to ensure an understanding and alignment with the risk-based principles needed. This has been and will be an area that needs attention from the validation team.
As a project moves closer to the next step in the life cycle, Process Validation (referred to as Process Performance Qualification in the latest FDA guideline), the state of the Quality Management System becomes much more important. This represents both a practical and cultural change for the Chinese pharma manufacturing industry. Quality Management Systems have traditionally been focusing on how to achieve the product specification in accordance with GMP and Pharmacopeia by extensive end testing of products and intermediates. Process Validation has frequently consisted of three batches, tested in accordance with the product specification as defined in the registration file. There is no scientific basis for specifying the number of validation batches to three, and while this practice may still be acceptable in the EU, there is a movement in the US on to a more science-based approach. Extended testing i.e extra tests outside the product specification to obtain an even more robust verification, an expected practice in the EU and US has not been the normal practice in China either.
One of the main advantages of risk and science-based validation is that it provides a control strategy based on residual risk after process design and equipment qualification. This control strategy is an input for the Design of Experiments for process validation batches that provides a scientific rationale for the number of batches and defines the extended testing needed. Control strategies also help define appropriate programmes for continuous process performance monitoring i.e. Continued Process Verification. In order to achieve this goal there is a need to implement a Pharmaceutical Quality System including a number of elements that traditionally have not been prioritised. Change Management, Deviation Management, Supplier Management, Continual Improvements and Product Quality Reviews are examples of activities that need to be implemented not just in the documentation, but also in practice. This is a major cultural change for most Chinese pharma manufacturers.
Currently the industry in China is waiting to see how the Chinese FDA will interpret and enforce the new GMPs from 2011. This may seem to be long overdue since now over three years have passed since the regulation was launched, but the CFDA recognised that the new requirements were setting a completely new standard for the industry and gave a grace period until January 2014. This year a large number of sterile and aseptic manufacturers are being inspected by the authorities. It has been recognised that while central CFDA has been very strict in their interpretation, China is an enormous country and it will take time for the new interpretations to trickle down to the provincial CFDA inspectorates. Therefore, the industry is still awaiting the outcome of local inspections. This gridlock will probably remain for the rest of 2014 and well into 2015. If the CFDA is enforcing the new GMP as vigorously as they have announced, there will surely be an increasing demand for new equipment and facilities, but the main demand will be new well-documented and improved Quality Management Systems.
Many of the leading manufacturers are not waiting for the results of the first real CFDA new GMP audits, but have instead chosen to aim for compliance with EU GMPs for their current project portfolio. This might even be the case for projects without objectives of exporting to the EU, but is rather done to be able to set up strict quality objectives for their projects.
Conclusions
Our recent experience suggests that leading Chinese manufacturers not only have adopted the Quality by Design approach, but that they also fully appreciate the regulatory implications as well as the business drivers in implementing enhanced process understanding.
In applying Quality by Design and Risk and Science-based approaches, the Chinese are in some regards better equipped to manage the changes of the organisations that this brings, due to the lack of quality history. Since there is a lack of understanding regarding the extent to which the new CFDA GMP will be enforced, and the similarities on paper between EU and the new Chinese GMPs, many leading manufacturers aim for compliance with EU GMP in current projects. While the industry as a whole is still behind EU and the US in terms of cGMP compliance, the gap is decreasing, and it is decreasing rapidly. In a not too distant future, Chinese manufacturers are likely to catch up and even surpass EU and US manufacturers both in terms of compliance and quality performance. However, China still has issues with management, leadership, innovation and creativity that are slowing the pace of the development down for the time being. Changing this probably presents a bigger challenge than implementing new industry regulations and guidelines.
-- Issue 21 --












