
For many of us in pharma and biotech R&D trained as academic scientists, having timelines associated with drug discovery and development projects can feel both unintuitive and bizarre at times. This is science, we find ourselves thinking. How can it fit on a GANTT chart?
Learning to corral the experimental vagaries of drug discovery and development into a timeline and a budget is a critical skill, both for an individual project lead’s career and for a company’s success. In this article, we’ll dig into how that’s even possible. To make things a bit concrete, we’ll use a specific scenario (small-molecule preclinical development for Oncology), but the general guidelines here are readily applied to nearly all therapeutic areas and modalities. Similar approaches can be used for more efficient and cost-effective screening, lead optimisation or early clinical development as well.
So, let’s take a closer look at what has to happen when a molecule is in the preclinical development stage. Typically, the project team has an identified molecule in hand and now has to put together the IND-enabling package, which gates the entry of the molecule into the clinic. For the pharmacologist, this boils down to answering a number of questions that allow for the design of a science-driven Phase I trial:
• What is the right starting dose for the candidate drug?
• What is the dose escalation scheme?
• What is the right dose route and schedule?
• What are the right time points and dose levels for pharmacodynamic (PD) biomarker collection?
• Has the patient population been identified already, or will it be identified during Phase I?
These questions are critical for the IND package, because they will define the clinical strategy, and the right (or wrong) answers can be decisive for a programme’s fate.
These questions are also deceptively easy to answer, however, because they can often be brushed off with an opinion. Because there is a tendency to seek certainty as early in development as possible, there is a tendency for project teams to shoot from the hip, creating an artificial sense of progress by locking down answers to these questions with very little preliminary workup. Unfortunately, a lack of rigour in addressing these questions will usually lead to fatal consequences for a programme in early clinical development (see this article of mine for more details on that!).
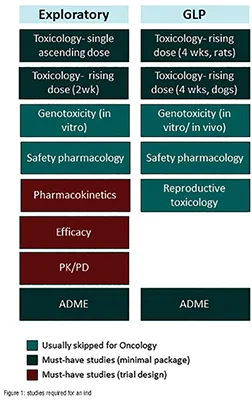
Part of the issue lies with the approach taken in preparing for the IND application. Often, teams will work their way through a laundry list of studies that looks something like Figure 1. The problem with the “laundry list” approach is that a project team can easily execute on the studies without having them answer the questions above.
It takes careful planning to address the questions above, and often project teams can end up thrashing around without getting to the ‘killer’ slide necessary to support a data-driven choice for each one. A better approach is to frame each clinical translation question in terms of a scientific hypothesis and then try to address that with a set of experiments. Easier said than done, right? Let’s dig into how to make this happen in practical terms.
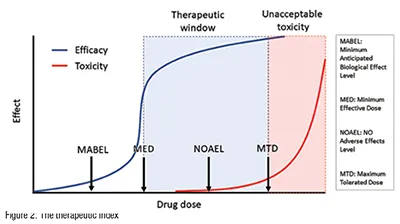
At the heart of the development paradigm for a successful oncology drug lies the therapeutic window- the gap between the efficacious and the toxic dose (Figure 2). (A closely related concept, the therapeutic index, is the ratio of the efficacious dose to the toxic dose). In oncology, for example, drugs that have a therapeutic index of one or more are likely to succeed in the clinic. Drugs with a therapeutic index of less than one will fail. (Every time. It’s as simple as that.)
So, it goes without saying that estimating the therapeutic index efficiently is the key to advancing molecules to the clinic. Many, if not most, design choices during preclinical development impact the therapeutic index- choice of formulation, dose schedule, dose route, dose escalation scheme and patient population, to name a few. If a molecule doesn’t have a therapeutic index of greater than one on the intended development path, then the team can iteratively refine the development path (changing one or more of the parameters mentioned above) to seek a wider therapeutic index. Ironically, many teams avoid making a rigorous assessment of a programme’s therapeutic index, as they are afraid of the consequences of the wrong answer. In reality, early and rigorous therapeutic window assessments maximise the chances of programme success.
The therapeutic window assessment also underpins many of the key questions for clinical trial design. Each of the critical questions above can be framed and answered rigorously, if you have the dose-response curves for efficacy and toxicity in hand:
What is the right starting dose for the candidate drug? Project the dosetoxicity relationship for humans, setting the starting dose based on toxicology (based on the no adverse effect level) or on PD (Minimal Anticipated Biological Effect Level).
What is the dose escalation scheme? Based on the steepness of the projected dose-toxicity relationship, decide whether a traditional 3+3 scheme is acceptable (you can usually do better). If not, use the projected dose-toxicity relationship as a Bayesian prior and design a dynamic dose escalation scheme that leverages this prior to escalate quickly and safely.
What is the right dose route and schedule? Estimate the pharmacokinetic (PK) parameter that is best correlated with efficacy (the PK “driver” of efficacy). Do the same for toxicity. Leverage this knowledge in identifying the dose route and schedule that provide the greatest therapeutic window. For example, if peak concentration (Cmax) drives toxicity, while total drug concentration over time (AUC, or area under the curve) drives efficacy, a once-weekly i.v. infusion may be a better idea than a single i.v. bolus delivered every three weeks. A difficult side effect profile will make it harder to achieve the full potential of the drug and will negate the added convenience of the single i.v. bolus dose.
What are the right time points and dose levels for PD biomarker collection? Build a similar PK/PD dose-response relationship for the biomarker(s). Leverage this PK/PD relationship to project the PD response over time in humans, by accounting for differences in PK between preclinical model species and humans.
Has the patient population been identified already, or will it be identified during Phase I? If the patient population has already been identified, construct PK/ efficacy dose-response relationships in a range of translationally relevant animal models. (Usually, for Oncology, this can be done in patient-derived xenograft models, which have very high predictive. value for this sort of work). If the patient population hasn’t been identified, you can use the projected dose-response relationship for efficacy as a Bayesian prior in a retrospective analysis at the end of a Phase I/II trial to zero in on sensitive subpopulations. The plans for this should ideally be in place before the start of the Phase I trial.
This is a high-level overview of some very complex topics, so there’s lots more to say on each of the above points. (Check out this article by me for a little more detail, or head over to our webpage for white papers and peer-reviewed publications by us on these topics).
With that said, it should be clear at the thirty-thousand foot level that the therapeutic window forms the key to efficient preclinical development, and most of the critical-path questions on the road to the IND are dependent on this analysis. Estimating the therapeutic window requires us to project the doseresponse relationship for efficacy and for toxicity in the clinical setting. Here’s the funny thing- getting to a precise quantitative estimate of the therapeutic window is much easier said than done!
Let’s use a simulated in vivo efficacy study to explain why this is so tricky. For oncology in vivo pharmacology, the workhorse model is the mouse xenograft. Mice are injected in their flank with cancer cells, these cells grow after about a week or two into small tumours, which are then either allowed to grow untreated or are treated over the study period. The impact on tumour growth is typically expressed as the ratio of treated/control tumour volume at the end of the study period (although there are better ways to do this, as this publication by us shows).
So, when running an in vivo efficacy study, you will start with an idea of what the maximum tolerated dose (MTD) of the drug is. Now, in a typical setting, you might do an initial efficacy study looking at the efficacy at MTD, ¾ MTD, ½ MTD and so on. The problem is that the relevant range for the dose-efficacy relationship can take many iterations to discover. In this simulated example, the underlying dose-efficacy relationship is steep. So, you run your first efficacy experiment starting from the MTD (40 mg/kg) and stepping down in ¼ intervals (Fig 3A). That didn’t go so well- it’s not a dose-response! So you try again, this time from 20 to 40mg/kg (Fig 3B)- still not a dose-response. Finally, you get it right on the third try (Fig 3C). This third run gives you the information you need to lock in the EC50 for the dose-response relationship for efficacy. But, unfortunately, each in vivo study cost (say) $65k, and took a month for the turnaround (optimistically). So that was ~$200k and 3 months of work.
Was there a better way? Yes, there was. Establishing the PK/PD relationship (which is also used in the IND) first with a mechanistically relevant biomarker (e.g. target occupancy) would have indicated that target occupancy saturates around 35 mg/kg, and most of the information is contained in the range between 28 and 35 mg/kg. A single PK/PD study (~$40k and 1 week of work if the assays are already in place), followed by a focused efficacy study would have gotten you the doseefficacy relationship for a total tab of ~$100k and one-and-a-half months. (Of course, this is not an apples-to-apples comparison because the PK/PD workup would need to be performed either way.) The big difference is this- using a modelbased approach gets you there reliably, while the trial-and-error method comes with open-ended timelines and budgets.
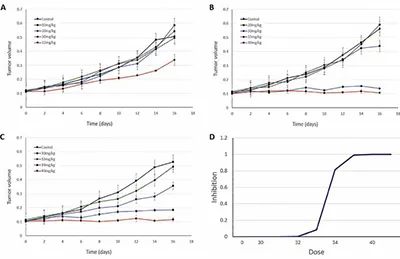
This is a simplistic simulation-based example, but it reflects what I’ve seen in my decades of experience as a preclinical and translational pharmacologist: using PK/PD modelling allows for more focused experimental designs that can limit the ‘thrashing around’ that is so common in preclinical development.
A rigorous approach to preclinical development requires three things:
• commitment to focusing on the critical-path questions
• persistence in getting the data required to answer these questions quantitatively
• judicious application of PK/PD modelling to narrow down the search space and speed things up.
The end products are an IND application and a Phase I design that are both scientifically “tight” and cost-effective. While we focused here on small-molecule oncology, these approaches can be applied to almost any therapeutic area and modality. The details may change (for example, in other therapeutic areas the focus can end up being on the relationship between target occupancy and drug biology, rather than therapeutic index per se), but the strategy is almost identical.
In lean times, the ability to execute reliably on time and under budget can make the difference between life and death for early-stage biotechs (and projects). Model-based pharmacology allows companies to derisk the road to the IND by allowing them to focus limited resources on the questions that matter most. This focus allows for a more robust and rigorous package and leaves room for recovery from unexpected developments on the way there.
This was a quick overview of a complex topic - there’s much more that can be said on many of the aspects covered here. Head over to our website for more white papers and publications, or feel free to ping me directly on LinkedIn if you have more questions!








