
Nanomedicines refer to drugs, medical devices and health products developed using nanotechnology with the aim of diagnosing, monitoring and treating diseases at the molecular level. Nanomedicines can be used to achieve the same therapeutic effect at smaller doses than their conventional counterparts due to their nano size, which improves their solubility and bioavailability. Broadly, there are three types of nanomedicines namely nanocarriers for drug delivery, nanosuspensions and nanoparticles for bioimaging. Though nanomedicines possess tremendous potential in the treatment and diagnosis, in-spite there are concerns regarding their safety and toxicity? Therefore, regulatory framework or guidelines for nanotechnology products must be in place for extending the immense positive benefits of nanomedicines to the society.
Regulatory scenario
So far, neither engineered nanoparticles nor the products and materials that contain them are subject to any special regulation regarding production, handling or labeling. Regulatory bodies such as the United States Environment Protection Agency (EPA) and US Food and Drug Administration (FDA) or the Health and Consumer Protection Directorate of the European Commission have taken initiatives to deal with the potential risks posed by the nanoparticles. Berkeley, California is currently the only city in the United States to regulate nanotechnology through an Assembly Bill, 2006.
On the basis of Nanotechnology Task Force report in 2007, the FDA issued its first draft guidance in June 2011 titled as Considering Whether an FDA-Regulated Product Involves the Application of Nanotechnology: Draft Guidance for Industry. Till date, FDA made available only the draft guidance documents but has not yet established specific formal regulatory guidelines for any nano-product. To promote nanotechnology, Government of India launched a Mission on Nano Science and Technology (i.e. Nano Mission) in May 2007 which is a successor of Nano Science and Technology Initiative (NSTI) of Department of Science and Technology (DST), 2001 but no guidelines are drafted for regulating nano-products in general.
Regulatory challenges
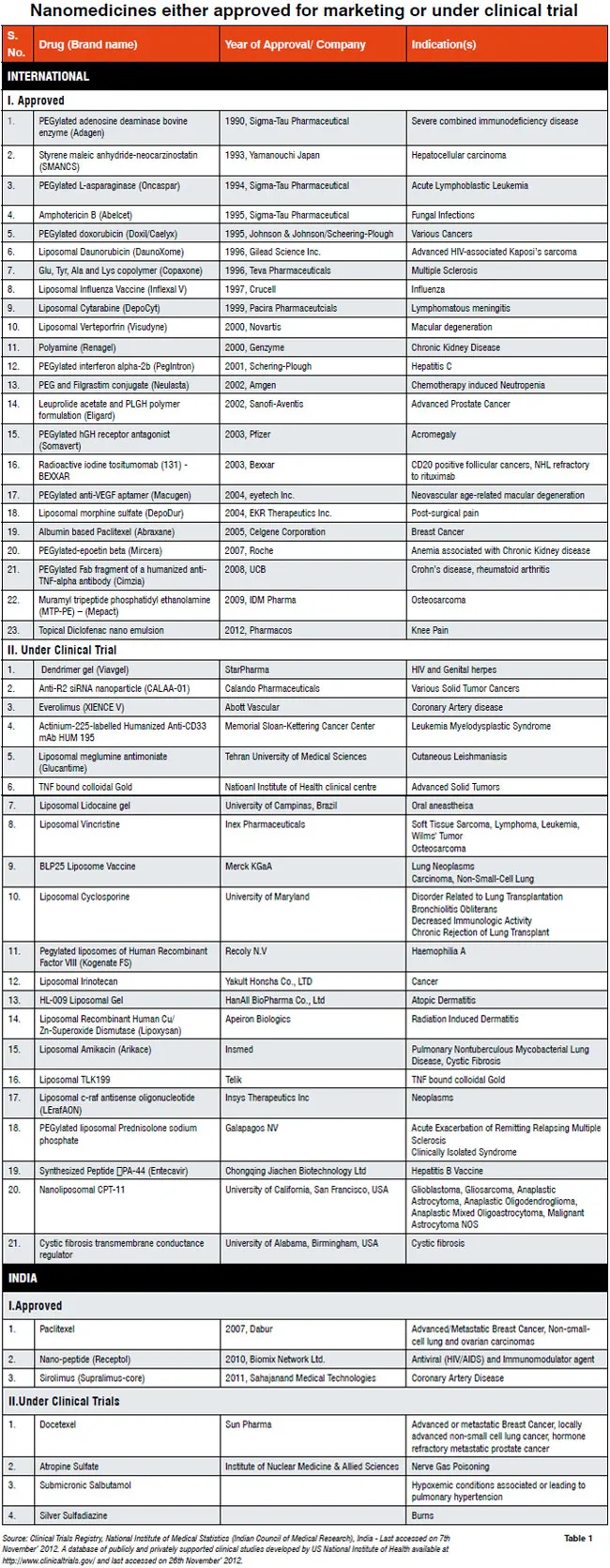
The nanomedicines either approved for marketing or under clinical trial are listed in Table 1. Currently under FDA, 591 clinical trials are going on for various liposomal preparations, which is just one type of a nanomedicine; while in India, the total number of nanomedicines under clinical trial is 21. In India, nanomedicines are slowly appearing in the market, for example, Ranbaxys Volini nanogel; therefore, regulatory guidelines are required to mitigate the hazardous outcome of any nanotechnology-based drug. The guideline preparation for nanomedicines is certainly not an easy task due to the lack of extensive and deep scientific knowledge as well as tools and techniques. There are few scientific and analytical concerns in the context of regulatory guideline for nanomedicines are discussed here.
Firstly, the definition of nanoproducts is not universally accepted and as a result, there is no homogenisation of the acceptable limit, for example, the US National Nanotechnology Initiative (NNI) launched in 2000 considers the dimension from 1-100nm. The U.K. Royal Society and Royal Academy of Engineering, 2004 proposed a range of 0.2-100nm. The Friends of the Earth Australia recommend defining nanoparticles up to 300nm in size. The definition of nanoparticles is important as their size has proportionate increase in surface to volume ratio, admissible changes in physical, pharmacokinetic & pharmacodynamic properties, toxicity and biosafety level, direct or indirect environmental and ecosystem impact.
Secondly, the issues related to the production of nanoparticles such as the infrastructure, training of human resource for handling the nano raw and manufactured materials, occupational hazards and associated health risks, quality control and their assessment in the absence of adequate technology for analysis
Thirdly, the aspects related to the clinical usage of nanomedicines as they are supposed to deliver the drugs locally in high doses at a particular cellular site. This mechanism of drug delivery requires extensive safety data at least up to pre-clinical stage before market approval. The localised high drug dose might lead to wide arrange of possible adverse effects, which may be aggravated as a result of improper distribution and excretion of nanomedicines. The localised high drug dose may lead to toxicity of a particular cell/organ type (lethal particularly in patients like diabetes and chronic kidney disease), or emergence of antibiotic resistant superbugs in case of antibiotics, hypersensitive reactions as a result of either interference of cellular processes due to retention of nano particulate metallic / nonmetallic vehicles or generation of antibodies against the antibodies used to direct a nanomedicine towards the target. Furthermore, due to the nano size of these medicines, they possess exceptional mobility quality; as a result, they may cross Blood Brain Barrier. The unwanted presence of nanomedicines may compromise the ability of the brain either severely or for a long term. Also, as a result of their altered biochemical activity, they may bind either with key cellular protein(s) or unwanted molecules such as toxins, and resultantly, interfere with the protein action or enhance the toxic effect of toxins.
Fourthly, niche area of detection protocols and methodologies for nanomedicines needs to be encouraged. The existing methods may provide both the qualitative and quantitative data but will not be sufficient to probe the specific aspects such as counting and assessing the homogeneity of nanoparticles in a drug formulation especially nano-suspension, quantitative assessment related to the retention / excretion of both metallic and biological nano-vehicles such as gold particles, carbon nanotubes and liposomes in human system and their risk assessment.
Fifth, nanomedicines pharmacokinetics have deviated from the normal course. As a result they are bioavailable for a sustained period, and, therefore, for such drugs the over-the-counter approach may lead to potentially increased health hazard to the public. Therefore, the regulatory bodies needs to assess whether the nanomedicines are allowed to market under the strict medical supervision or simply labeled as Nano. It is in fact not easy to reach either of these conclusions in the absence of availability of significant toxicity, biosafety data analysis as well as their environmental risk assessment.
Lastly, delegation of responsibility to the appropriate group of government organisations is desired for formulating draft guidelines for nanomedicines through a wide array of consultative process involving various stakeholders such as sndustries, academia, clinicians and end users. Furthermore, there is immediate need for establishing world class regulatory laboratories and testing facilities at least at the federal level. Simultaneously, risk assessment expertise in terms of personnel, guidelines and technical standards needs to be buildup.
Conclusion
Nanotechnology is highly inter-disciplinarily in nature, which requires a diverse set of stakeholders and their coordinated efforts. Limitations such as inadequate knowledge regarding nanoparticle behaviour, absence of standardised nomenclature, test methods, and characterisation of nanoparticles, shortage of trained personnel are some of reasons which are possibly hindering the process of determining primary jurisdiction for combination products, nanomedicine-specific safety protocol, ineffective control of nanoparticle and developing the framework or pathway for controlling the manufacturing processes, product quality and safety of nanomedicines.
Authors Note: The views expressed are personal and has NO relation with official position in Department of Biotechnology, Ministry of Science & Technology, New Delhi, India.
-- Issue 19 --





